
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRepeat proteins as versatile scaffolds for arrays of redox-active FeS clusters†
Sara H.
Mejias‡
 ac,
Zahra
Bahrami-Dizicheh‡
b,
Mantas
Liutkus
a,
Dayn Joshep
Sommer
b,
Andrei
Astashkin
d,
Gerdenis
Kodis
b,
Giovanna
Ghirlanda
*b and
Aitziber L.
Cortajarena
*ace
ac,
Zahra
Bahrami-Dizicheh‡
b,
Mantas
Liutkus
a,
Dayn Joshep
Sommer
b,
Andrei
Astashkin
d,
Gerdenis
Kodis
b,
Giovanna
Ghirlanda
*b and
Aitziber L.
Cortajarena
*ace
aCIC biomaGUNE Paseo de Miramón 182, E-20014 Donostia-San Sebastian, Spain
bSchool of Molecular Sciences, Arizona State University Tempe, AZ 85287-1604, USA. E-mail: giovanna.ghirlanda@asu.edu
cIMDEA-Nanociencia and Nanobiotechnology Unit Associated (CNB-CSIC), Campus de Cantoblanco, E-28049 Madrid, Spain. E-mail: alcortajarena@cicbiomagune.es
dDepartment of Chemistry and Biochemistry, University of Arizona, Tucson, AZ 85271, USA
eIkerbasque Basque Foundation for Science, E-48011 Bilbao, Spain
First published on 4th March 2019
Abstract
Arrays of one, two and four electron-transfer active [4Fe–4S] clusters were constructed on modular tetratricopeptide repeat protein scaffolds, with the number of clusters determined solely by the size of the scaffold. The constructs show reversible redox activity and transient charge stabilization necessary to facilitate charge transfer.
Molecular materials for solar energy harvesting, molecular electronics and nanoscale devices require systems capable of facilitating long-range electron transfer (ET) with high quantum yield.1,2 As biological ET becomes better understood, nature-inspired ET systems are emerging as alternatives to purely synthetic designs. In nature, medium and long distance electron transfer is often performed by arrays of cofactors held at specific distances within complex proteins.3–5 Fe–S clusters are some of the most common cofactors utilized for this purpose. Cubane-like [4Fe–4S] clusters are the most abundant type of Fe–S clusters found in protein structures, involved in many natural ET processes (e.g. photosynthesis, respiration).3,5–8 A common coordination motif utilizes four conserved cysteines coordinated to the cluster and ensconced in the hydrophobic core of the protein.9–15 Most synthetic models aimed towards engineering of functional redox enzymes have been designed to bind a single electronically isolated cluster in the protein scaffold or in the proximity of a catalytically active cofactor.9–11 Recently, first examples of two [4Fe–4S] clusters incorporated at design-determined distances in the hydrophobic core of a three-helix bundle have been reported.12,13 However, designing multi-center Fe–S proteins remains a challenge.
Modular assembly of multi-center ET-active systems based on repeat proteins was identified as a viable strategy. The structural simplicity and intrinsic modularity of the repeat proteins allow to use them as simple units for bottom-up fabrication, where each repeat unit can be used as a building block with individually engineered properties and functionalities.16,17 Consensus tetratricopeptide repeat (CTPR) protein, composed of helix–turn–helix motifs that connect in a sequence to form a right-handed superhelical structure,18 possesses the robustness and stability needed to support supramolecular assemblies19–21 or act as support structures in solution and solid state.22–24 In the current work, a four-sulfur coordination site was ‘grafted’ onto CTPR protein scaffold, forming arrays of functional ET-active [Fe–S] clusters with precisely defined distance and orientation. The system establishes a foundation for the modular design of long-range ET conduits. To the best of our knowledge, this is the first example of a single scaffold used to assemble a controlled number of clusters.
Based on the crystal structure of tryptophanyl-tRNA synthethase (PDB ID: 2G36) and on a previous design of a [4Fe–4S] cluster-coordinating peptide,12,13 a four-cysteine [4Fe-4S] coordination site was modeled at the interface of two adjacent CTPR repeats (PDB: 2HYZ), on the concave surface of CTPR superhelix. Of the several possible positions for the cysteines, the design that supported the correct distances and rotamers for the four coordinating cysteines, Y5C and N9C in the first repeat and E2C and N6C in the second repeat, was selected. A [4Fe–4S] cluster was then manually docked into the designed binding pocket using PyMOL software and the resulting model was subjected to three rounds of energy minimization using GROMACS, with iterations of manual modeling. The side chain conformations and backbone geometry of the designed cysteines were compatible with the TPR helical fold and with the cluster coordination distances observed in natural coordination sites. The two-repeat cluster-forming unit was sequentially aligned with consecutive sections of an eight-repeat model to provide models of two- and four-cluster assemblies. The optimized models show no global structural deviations from the crystal structure of CTPR protein. Based on the models, the clusters in the multi-cluster constructs are about 16 Å apart (Fig. 1).
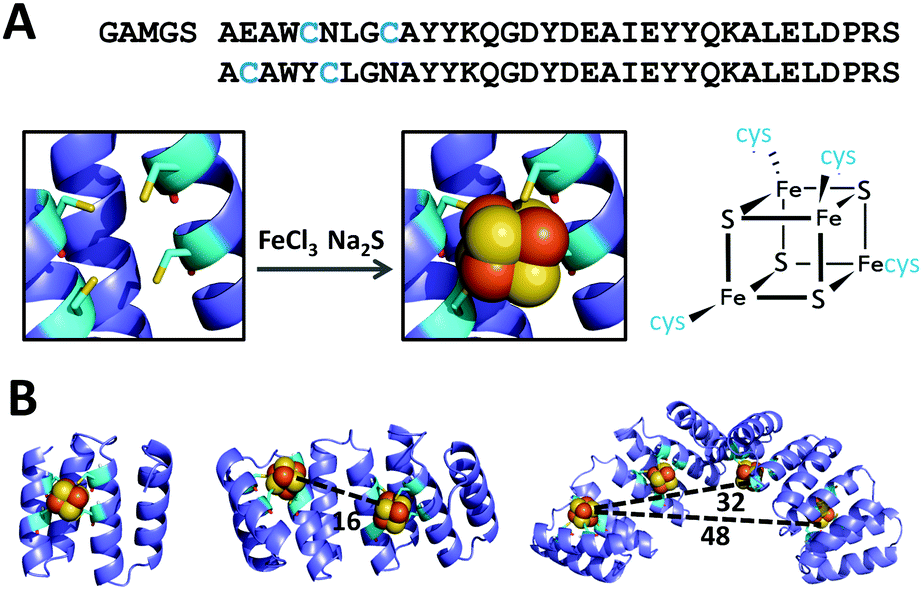 | ||
| Fig. 1 Design of CTPR proteins for the incorporation of [4Fe–4S] clusters. (A) Top: Sequence of modified CTPR2 cluster binding unit; residues mutated to cysteines are highlighted in blue: Y5C and N9C in the first repeat and E2C and N6C in the second repeat. Bottom: Structural model of the designed 4-Cys site (left) and the [4Fe–4S] cluster (right). (B) Models of CTPR2-[4Fe–4S], CTPR4-2[4Fe–4S], and CTPR8-4[4Fe–4S], containing 1, 2 and 4 [4Fe–4S] clusters, respectively. Distances listed in angstroms. | ||
Cubane-type [4Fe–4S] clusters within the proteins were formed using an established in situ synthetic procedure from iron(III) chloride and sodium sulfide under anaerobic conditions.25 The UV-vis spectra of the resulting CTPR2-[4Fe–4S], CTPR4-2[4Fe–4S] and CTPR8-4[4Fe–4S] constructs (Fig. S1, ESI†) show broad absorption peaks at 345, 430 and 630 nm, characteristic of charge transfer excitations from sulfur to iron in [4Fe–4S]2+ clusters,26 but red-shifted compared with inorganic [4Fe–4S] clusters due to the hydrophobic environment of the protein.10,26,27 The absorption at 430 nm disappeared upon reduction of the clusters to [4Fe–4S]+ state with sodium dithionite, as expected for cuboidal [4Fe–4S] clusters.28–30
Iron content (measured using the ferrozine method31) in the protein complexes (determined using Bradford assay32,33) was in agreement with a single [4Fe–4S] cluster per binding cavity (Table 1): one cluster in CTPR2 scaffold, two clusters in CTPR4 and four clusters in CTPR8. Circular dichroism (CD) analysis showed the CTPR proteins retained α-helical structure in all three constructs, indicating that the incorporation of the [4Fe–4S] clusters did not affect the structure of the protein scaffolds (Fig. S2, ESI†). To the contrary, thermal denaturation analysis showed that the clusters increased global stability of the proteins, with the effect most pronounced for the 2-repeat protein, which cannot benefit from the stabilizing effect of the extended superhelical structure due to its size (Fig. S3, ESI†). The Tm values for CTPR2, CTPR4 and CTPR8 were 55, 68, and 75 °C, respectively, while the respective Tm values for CTPR2-[4Fe–4S], CTPR4-2[4Fe–4S] and CTPR8-4[4Fe–4S] were 65, 73 and 75 °C. This stabilizing effect, arising from a rigid [4Fe–4S] cluster holding protein helical fragments in place, is in line with previous results.12,13
| Construct | Fe atoms per protein chain |
|---|---|
| CTPR2-[4Fe–4S] | 4.60 ± 0.38 |
| CTPR4-2[4Fe–4S] | 7.62 ± 0.22 |
| CTPR8-4[4Fe–4S] | 16.44 ± 1.33 |
Electron paramagnetic resonance (EPR) spectroscopy confirmed the iron–sulfur clusters initially formed in an EPR-silent [4Fe–4S]2+ resting state. The lack of signal further confirmed cluster identity, ruling out other possible clusters (e.g. [3Fe–4S]) that are EPR active in the resting state. Reduction with sodium dithionite produced the EPR active [4Fe–4S]1+ state: all three constructs displayed nearly identical spectra with principal g-values of 1.88, 1.93, and 2.06 (Fig. 2), typical of [4Fe–4S]1+ clusters.34 Features indicative of exchange and/or magnetic dipole interactions between the clusters, observed with natural ferredoxins containing two [4Fe–4S]1+ clusters at 10–15 Å from each other,35 were not detected for CTPR4-2[4Fe–4S] and CTPR8-4[4Fe–4S] constructs, suggesting lack of sufficiently strong interactions between the clusters.
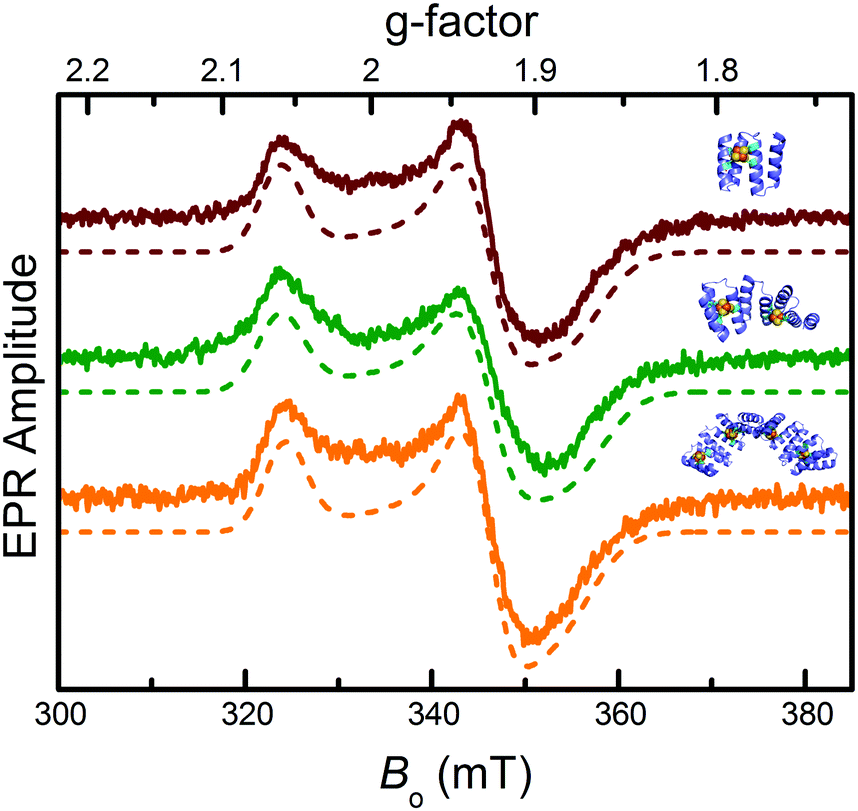 | ||
| Fig. 2 EPR characterization of the CTPR-bound [4Fe–4S] clusters. From top to bottom: EPR spectra of sodium dithionite-reduced CTPR2-[4Fe–4S] (red), CTPR4-2[4Fe–4S] (green) and CTPR8-4[4Fe–4S] (orange) constructs (solid lines) and numerically simulated EPR spectra (dotted lines). Simulation parameters provided in ESI.† | ||
Square wave voltammetry was used to assess the redox potential of the clusters. Electrochemically quasi-reversible signals were observed for all three constructs, with redox potentials of ca. −0.21 V, −0.24 V and −0.24 V vs. SCE for CTPR2-[4Fe–4S], CTPR4-2[4Fe–4S], and CTPR8-4[4Fe–4S], respectively (Fig. 3 and Table S1, ESI†), and peak to peak separation of about 250 mV, indicative of moderate electron transfer rate between the electrode and proteins.36 These values fall within the range expected for the relatively solvent exposed low-potential [4Fe–4S] clusters.26,27,37 The similar behavior of the three constructs suggests the clusters react mostly independently, with little interaction between neighboring clusters within the larger assemblies. At constant protein concentration, the current was proportional to the number of clusters in the constructs, as expected (Fig. S4, ESI†). However, the larger constructs showed increasingly more negative redox potentials, despite identical environment around the clusters. This trend could be attributed to the difficulty of successive cluster reduction after a first cluster in a construct has been reduced, thus suggesting at least some degree of interaction between the clusters. Structural factors were then considered as a reason for the lack of charge transfer between the clusters. Transient absorption (TA) spectroscopy was thus employed to investigate charge transfer pathways of the clusters.
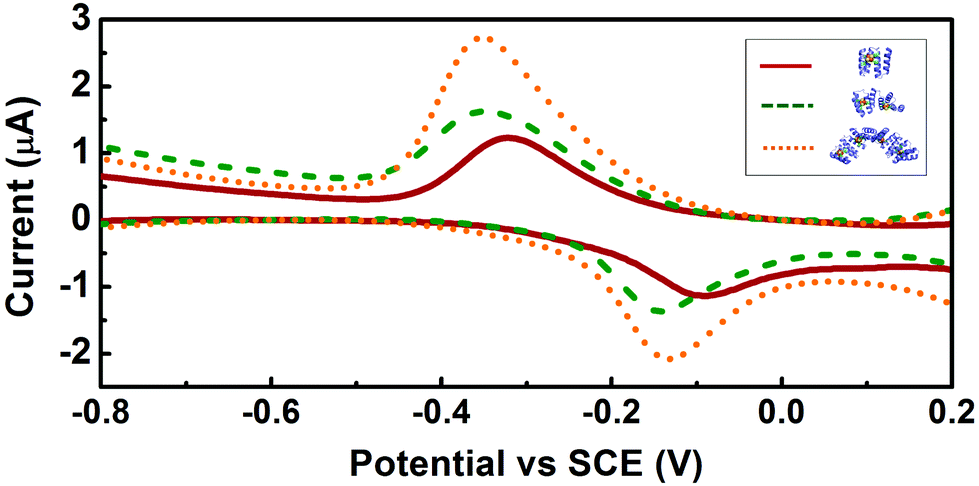 | ||
| Fig. 3 Square wave voltammetry of the CTPR-[4Fe–4S] complexes. Voltammograms of CTPR2-[4Fe–4S], CTPR4-2[4Fe–4S] and CTPR8-4[4Fe–4S] are shown in red, green and orange, respectively. | ||
Laser pulses of ∼100 fs at 400 nm were used to promote ligand to metal charge transfer (LMCT) transitions from sulfur to iron atoms.38 Global analysis of the transient absorption data gave three evolution-associated-difference spectra (EADS).39 EADS from all three proteins have similar features and the TA was satisfactorily fitted with almost identical lifetimes (Fig. 4A and Fig. S5, ESI†), indicating at least 3 transient states (species) involved in the photo induced charge transfer process, but no direct electron transfer between the [4Fe–4S] clusters in the protein. However, the EADS showed that the 400 nm laser pump induces an internal reduction of the [4Fe–4S] cluster, and the surrounding protein is capable of stabilizing that state via long-range redox reaction to its outside, creating a long-lived (about 7 ns) charge-separated state. As such, the EADS corresponding to the species with 0.4 ps lifetime shows broad ground state bleaching (GSB) with maxima below 460 nm and around 600 nm (Fig. 4A and Fig. S5, ESI†), characteristic of the S(3p) → Fe(3d) LMCT excited state38 that, due to the short lifetime, may be attributed to the transient internal electron transfer (ET) from a bridging sulfide (ET pathway 1, Fig. 4B). Similarly, the 6 ps EADS show GSB maxima below 460 nm and around 650 nm. In this case, due to the longer lifetime and the red-shifted GSB, the most probable path involves a cysteinyl sulfur transferring an electron to the iron in the [4Fe–4S] cluster (ET pathway 2, Fig. 4B), thus generating an electron hole in the cysteine. Interestingly, this state decays in part by forming a transient state with 7.5 ns lifetime (7.5 ns EADS) and characteristic bleaching of tyrosyl or tryptophanyl radical around 510 nm.40 This suggests the electron hole on the cysteine is most likely quenched by proton coupled electron transfer (PCET) promoted by the linked tryptophan where the electron acceptor is the cysteine hole and the proton acceptor is possibly water (pathway 3, Fig. 4B).41 Cysteine-linked tyrosine might be involved in this PCET as well (pathway 3′), but shorter distance (fewer connecting bonds between chromophores) and bigger driving force for the electron transfer would probably favor the tryptophan.
 | ||
| Fig. 4 (A) EADS for the CTPR2-[4Fe–4S] from the global fit of TA data with three lifetimes: 0.4 ps (straight line), 6 ps (dashed line) and 7.5 ns (dotted line). The 7.5 ns EADS is magnified by a factor of 8 for better visibility. (B) Proposed electron transfer pathways. Arrows show the direction of electron transfer. Iron is in orange, sulfur is yellow, cysteines (Cys) are light blue, tyrosine (Tyr) and tryptophan (Trp) are green. | ||
This study has demonstrated the potential of CTPR proteins as a scaffold for [4Fe–4S] clusters, enabling to coordinate a desired number of redox active clusters in a single assembly, with the cluster count controlled solely through the size of the protein. The repeat nature of the protein also imposed a consistent distance of about 1.6 nm between the clusters in multi-cluster assemblies. The designed proteins coordinate the clusters with expected stoichiometry, with one cluster per engineered binding site, and with spectral characteristics similar to those observed in natural ferredoxins. The CTPR-[4Fe–4S] constructs remain well folded, with the characteristic helical structure of the TPR domain. The EPR spectra of the reduced protein-cluster complexes correspond to the [4Fe–4S]1+ state. Square wave voltammetry confirmed that the [4Fe–4S] clusters coordinated by the designed proteins are redox active and have redox potentials within the range of low-potential protein-bound clusters. Transient absorption spectroscopy showed that clusters form long-lived charge-separated states stabilized by the protein matrix, as observed in proteins involved in long range energy transfer. Direct interaction between the [4Fe–4S] clusters was not observed, but instead the excitation pathway was traced to the surface of the protein. Thus, the lack of cross-talk between the clusters is attributable to the structural factors of the system, in particular the large distance (>15 Å) between the clusters, the exposure of the clusters to the environment and the lack of driving force (e.g. difference in potentials) for inter-cluster communication, making the charge transfer to the environment (water) more favorable. Nevertheless, the reactivity of the clusters shows the potential of CTPR-[4Fe–4S] assemblies as a simplified platform for the studies of long-range ET, establishing the foundation for the modular design of long-range ET conduits.
Overall, this communication serves to further demonstrate the versatility of CTPR proteins as a platform for designing functional materials or assemblies with order maintained at a molecular level. CTPR proteins have proven to be robust scaffolds for complex multifunctional systems with diverse functional elements.22,23 The stabilization of redox active clusters brings the possibility of combining photo- and redox-active elements for full-fledged energy transfer conduits. As such, CTPR proteins show virtually unmatched potential as a protein scaffold for nanotechnological systems, including potential artificial photosystems.
This work has been supported by the European Research Council ERC-2014-CoG-648071-ProNANO (ALC), the Spanish Ministry of Economy and Competitiveness (MINECO) BIO2016-77367-C2-1-R (ALC), the Basque Government Elkartek KK-2017/00008 and the National Science Foundation (CHE-CLP award SusChEM 1508301). This work was performed under the Maria de Maeztu Units of Excellence Program from the Spanish State Research Agency – Grant No. MDM-2017-0720. SHM thanks the Basque Government for financial support through a PhD fellowship.
Conflicts of interest
There are no conflicts to declare.Notes and references
- P. Ceroni and V. Balzani, in The Exploration of Supramolecular Systems and Nanostructures by Photochemical Techniques, ed. P. Ceroni, Springer, Netherlands, Dordrecht, 2012, pp. 21–38 Search PubMed.
- S. R. Forrest and M. E. Thompson, Chem. Rev., 2007, 107, 923–925 CrossRef CAS.
- H. B. Gray and J. R. Winkler, Chem. Phys. Lett., 2009, 483, 1–9 CrossRef CAS PubMed.
- C. C. Moser, J. M. Keske, K. Warncke, R. S. Farid and P. L. Dutton, Nature, 1992, 355, 796–802 CrossRef CAS PubMed.
- P. Hinchliffe, Science, 2005, 309, 771–774 CrossRef CAS PubMed.
- J. C. Genereux and J. K. Barton, Chem. Rev., 2010, 110, 1642–1662 CrossRef CAS PubMed.
- J. L. Dempsey, J. R. Winkler and H. B. Gray, Chem. Rev., 2010, 110, 7024–7039 CrossRef CAS PubMed.
- L. A. Sazanov and P. Hinchliffe, Science, 2006, 311, 1430–1436 CrossRef CAS PubMed.
- M. L. Kennedy and B. R. Gibney, J. Am. Chem. Soc., 2002, 124, 6826–6827 CrossRef CAS PubMed.
- C. E. Laplaza and R. H. Holm, J. Am. Chem. Soc., 2001, 123, 10255–10264 CrossRef CAS PubMed.
- J. Grzyb, F. Xu, V. Nanda, R. Łuczkowska, E. Reijerse, W. Lubitz and D. Noy, Biochim. Biophys. Acta, Bioenerg., 2012, 1817, 1256–1262 CrossRef CAS PubMed.
- A. Roy, D. J. Sommer, R. A. Schmitz, C. L. Brown, D. Gust, A. Astashkin and G. Ghirlanda, J. Am. Chem. Soc., 2014, 136, 17343–17349 CrossRef CAS PubMed.
- A. Roy, I. Sarrou, M. D. Vaughn, A. V. Astashkin and G. Ghirlanda, Biochemistry, 2013, 52, 7586–7594 CrossRef CAS PubMed.
- J. Grzyb, F. Xu, L. Weiner, E. J. Reijerse, W. Lubitz, V. Nanda and D. Noy, Biochim. Biophys. Acta, Bioenerg., 2010, 1797, 406–413 CrossRef CAS PubMed.
- C. D. Coldren, H. W. Hellinga and J. P. Caradonna, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 6635–6640 CrossRef CAS.
- E. R. G. Main, J. J. Phillips and C. Millership, Biochem. Soc. Trans., 2013, 41, 1152–1158 CrossRef CAS PubMed.
- S. H. Mejias, A. Aires, P. Couleaud and A. L. Cortajarena, Advances in Experimental Medicine and Biology, 2016, pp. 61–81 Search PubMed.
- T. Kajander, A. L. Cortajarena, S. Mochrie and L. Regan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2007, 67, 800–811 CrossRef PubMed.
- S. H. Mejías, B. Sot, R. Guantes and A. L. Cortajarena, Nanoscale, 2014, 6, 10982–10988 RSC.
- S. H. Mejias, P. Couleaud, S. Casado, D. Granados, M. A. Garcia, J. M. Abad and A. L. Cortajarena, Colloids Surf., B, 2016, 141, 93–101 CrossRef CAS PubMed.
- T. Z. Grove, L. Regan and A. L. Cortajarena, J. R. Soc., Interface, 2013, 10, 20130051 CrossRef PubMed.
- S. H. Mejías, J. López-Andarias, T. Sakurai, S. Yoneda, K. P. Erazo, S. Seki, C. Atienza, N. Martín and A. L. Cortajarena, Chem. Sci., 2016, 7, 4842–4847 RSC.
- J. López-Andarias, S. H. Mejías, S. Tsuneaki, M. Wakana, S. Shu, F. Ferran, O. Sílvia, A. Carmen, M. Nazario and A. L. Cortajarena, Adv. Funct. Mater., 2017, 28, 1704031 CrossRef.
- P. Couleaud, S. Adan-Bermudez, A. Aires, S. H. Mejías, B. Sot, A. Somoza and A. L. Cortajarena, Biomacromolecules, 2015, 16, 3836–3844 CrossRef CAS PubMed.
- M. L. Antonkine, E. M. Maes, R. S. Czernuszewicz, C. Breitenstein, E. Bill, C. J. Falzone, R. Balasubramanian, C. Lubner, D. A. Bryant and J. H. Golbeck, Biochim. Biophys. Acta, Bioenerg., 2007, 1767, 712–724 CrossRef CAS PubMed.
- W. V. Sweeney and J. C. Rabinowitz, Annu. Rev. Biochem., 1980, 49, 139–161 CrossRef CAS PubMed.
- M. L. Antonkine, M. S. Koay, B. Epel, C. Breitenstein, O. Gopta, W. Gärtner, E. Bill and W. Lubitz, Biochim. Biophys. Acta, Bioenerg., 2009, 1787, 995–1008 CrossRef CAS PubMed.
- P. M. Vignais, B. Billoud and J. Meyer, FEMS Microbiol. Rev., 2001, 25, 455–501 CrossRef CAS PubMed.
- Z. Jin, M. Heinnickel, C. Krebs, G. Shen, J. H. Golbeck and D. A. Bryant, J. Biol. Chem., 2008, 283, 28426–28435 CrossRef CAS PubMed.
- A. Dubini, F. Mus, M. Seibert, A. R. Grossman and M. C. Posewitz, J. Biol. Chem., 2009, 284, 7201–7213 CrossRef CAS PubMed.
- P. Carter, Anal. Biochem., 1971, 40, 450–458 CrossRef CAS PubMed.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS PubMed.
- J. J. Sedmak and S. E. Grossberg, Anal. Biochem., 1977, 79, 544–552 CrossRef CAS PubMed.
- K. K. Eisenstein and J. H. Wang, J. Biol. Chem., 1969, 244, 1720–1728 CAS.
- R. Mathews, S. Charlton, R. H. Sands and G. Palmer, J. Biol. Chem., 1974, 249, 4326–4328 CAS.
- E. Hadzifejzovic, J. Stankovic, S. Firth, P. F. McMillan and D. J. Caruana, Phys. Chem. Chem. Phys., 2007, 9, 5335–5339 RSC.
- P. Hosseinzadeh and Y. Lu, Biochim. Biophys. Acta, Bioenerg., 2016, 1857, 557–581 CrossRef CAS PubMed.
- A. Aizman and D. A. Case, J. Am. Chem. Soc., 1982, 104, 3269–3279 CrossRef CAS.
- I. H. M. Van Stokkum, D. S. Larsen and R. Van Grondelle, Biochim. Biophys. Acta, Bioenerg., 2004, 1657, 82–104 CrossRef CAS PubMed.
- C. Aubert, P. Mathis, A. P. M. Eker and K. Brettel, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 5423–5427 CrossRef CAS.
- S. D. Glover, C. Jorge, L. Liang, K. G. Valentine, L. Hammarström and C. Tommos, J. Am. Chem. Soc., 2014, 136, 14039–14051 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: All synthetic and material characterization procedures, characterization data and models of the CTPR-[4Fe–4S] assemblies. See DOI: 10.1039/c8cc06827e |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2019 |