
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePoly(caprolactone-co-trimethylenecarbonate) urethane acrylate resins for digital light processing of bioresorbable tissue engineering implants†
Tobias
Kuhnt‡
a,
Ramiro
Marroquín García‡
 ab,
Sandra
Camarero-Espinosa
a,
Aylvin
Dias
c,
A. Tessa
ten Cate
de,
Clemens A.
van Blitterswijk
a,
Lorenzo
Moroni
*a and
Matthew B.
Baker
*a
ab,
Sandra
Camarero-Espinosa
a,
Aylvin
Dias
c,
A. Tessa
ten Cate
de,
Clemens A.
van Blitterswijk
a,
Lorenzo
Moroni
*a and
Matthew B.
Baker
*a
aDepartment of Complex Tissue Regeneration, MERLN Institute for Technology-Inspired Regenerative Medicine, Maastricht University, 6211 LK Maastricht, The Netherlands. E-mail: m.baker@maastrichtuniversity.nl; l.moroni@maastrichtuniversity.nl
bAachen-Maastricht Institute for BioBased Materials (AMIBM), Faculty of Science and Engineering, Maastricht University, The Netherlands
cDSM Materials Science Center, Urmonderbaan 22, 6167 RD, Geleen, The Netherlands
dTNO, P.O. Box 6235, 5600 HE Eindhoven, The Netherlands
eBrightlands Materials Center, Urmonderbaan 22, 6167 RD Geleen, The Netherlands
First published on 30th October 2019
Abstract
To exploit the usability of Digital Light Processing (DLP) in regenerative medicine, biodegradable, mechanically customizable and well-defined polyester urethane acrylate resins were synthesized based on poly(caprolactone-co-trimethlenecarbonate). By controlling the monomer ratio, the resultant fabricated constructs showed tunable mechanical properties, degradation and attached hMSC morphologies.
Additive manufacturing (AM) techniques allow the fabrication of complex three-dimensional (3D) objects with outstanding control in dimension and resolution.1,2 Among the different types of AM techniques, digital light processing (DLP) has emerged as an efficient, low-cost, and relatively fast technique for complex scaffolds like gyroids,3,4 vascular branches5 or for microarchitectures and microwells.6 Despite DLPs potential advantages, the lack of tunable biocompatible resins, in particular those with adequate bioresorbable and mechanical properties for tissue regeneration, have yet inhibited exploitation of this versatile photofabrication technique.7,8
DLP takes the advantage of a light source (UV or visible light) to induce a controlled photopolymerization reaction; thus, forming solid microarchitectures from liquid polymeric resins in a layer-by-layer approach.2 Generally, commercially available resins are based on monomers and short polymers containing acrylate or epoxy functionalities. Inherently, these polymers are sometimes biocompatible,9 however not necessary biodegradable.10 This need within the field has spurred recent studies on the creation of tunable and biodegradable DLP resins, for example poly(propylenefumarate) resins11–13 and thiol–ene based hydrogels.4,14
Light polymerizable resins are commonly acrylate functionalized polymers which are derived from poly(ε-caprolactone) (PCL),15 poly(lactic acid) (PLA),16 poly trimethylene carbonate (PTMC),17 or poly(propylene fumarate) (PPF).18 Previous work has developed such polymers for fabrication with stereolithography to yield biocompatible and biodegradable scaffolds. Such homopolymer-based resins have limited versatility in terms of mechanical properties and degradation rates, normally fine tuning the mechanical properties only by varying either the molecular weight or the crosslink density.19 In contrast to homopolymers, the use of co-polymeric resins that contain two or more polymers in the backbone have been proposed as a way to increase the range of mechanical properties of the final scaffold.3
Poly trimethylenecarbonate (PTMC) is an amorphous polymer, obtained by ring opening polymerization from the cyclic carbonate. Physically, PTMC exhibits a low glass transition temperature, ranging from −25 to −15 °C (depending on the molecular weight) and relatively low elastic modulus.20 Due to the relatively poor mechanical properties, PTMC is generally used in combination with semi-crystalline polymers such as PCL.21 The formed random PCL-co-PTMC polymer (PCT) offers advantages for biomedical applications due to the formation of non-acidic side products during biodegradation.22 Furthermore, these materials are characterized by a relatively low elastic modulus and ductile behavior.23
In this work, we studied the mechanical properties and accelerated degradation profiles of a series of PCT based photocurable resins. In total, 5 random PCT polymers (Mn: 4000 g mol−1) were synthesized and end-capped using acrylated L-lysine isocyanate (LDI-HEA) to form the final resins (Scheme 1). The reaction of LDI-HEA with the prepolymers introduces urethane moieties, which have the potential to increase the elastic modulus as well as enable other degradation pathways. Poly(urethanes) are widely used in biomedical devices due to their excellent mechanical response and inherent biocompatibility.24 The mechanical properties of cured samples were studied and accelerated degradation profiles were recorded for one month under basic conditions. Finally, a series of different scaffolds was printed using diphenyl(2,4,6-trimethylbenzoyl)phosphine oxide (TPO) as photo-initiator and 2-(2-ethoxyethoxy)ethyl acrylate (EOEOEA) as a reactive diluent to enhance the printability.
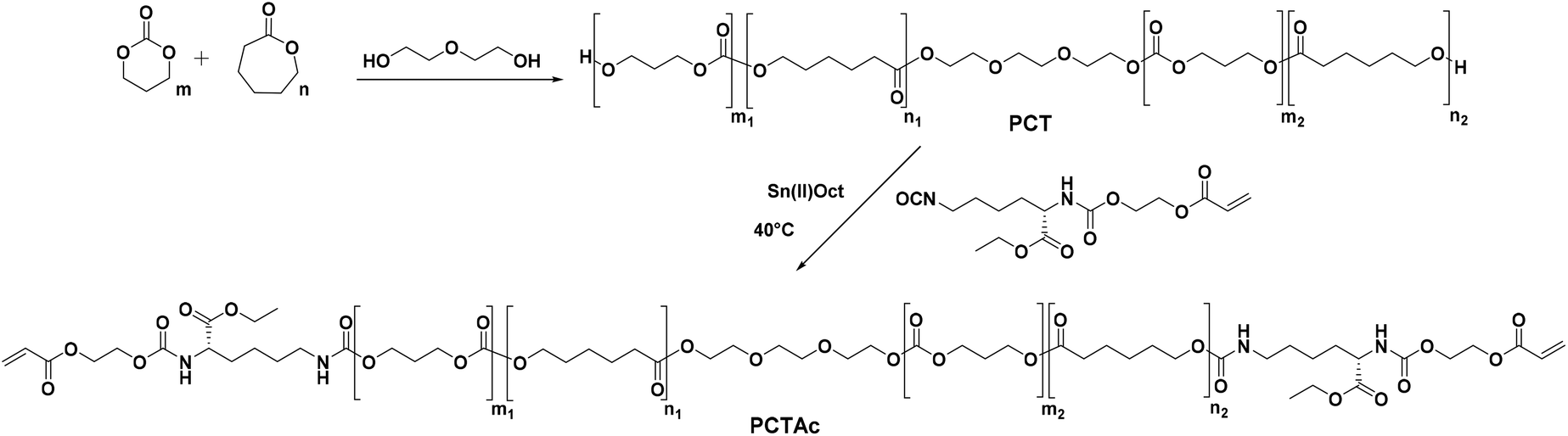 | ||
| Scheme 1 Synthetic route to PCTAc polymers based on PCT copolymerization followed by LDI-HEA endgroup functionalization. | ||
PCT prepolymers were synthesized via bulk ring opening polymerization of caprolactone (CL) and trimethylenecarbonate (TMC) using triethylene glycol (TEG) as initiator and Sn(II)Oct as catalyst in a ratio of 2000![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The prepolymers were characterized via1H and 13C-NMR spectroscopy and then assigned in conjunction with previous publications.25,26
:1. The prepolymers were characterized via1H and 13C-NMR spectroscopy and then assigned in conjunction with previous publications.25,26
A series of 5 copolymers was prepared. The monomer ratios were varied from (90:10, 75:25, 50:50, 25:75 and 10:90) CL to TMC. The monomer conversion after the polymerization was evaluated using 1H-NMR of the crude reaction mixture, the ratio between the monomer peaks to the polymeric CL and TMC signals (see ESI†). Fig. S8a† depicts the 1H-NMR spectra of the synthesized PCT prepolymer with a ratio 50:50 (CL:TMC), the region between 4.00 and 4.27 ppm was amplified to identify the methylene (–CH2–) corresponding to the CL and TMC units in the copolymer. The presence of extra peaks in the spectra was attributed to the formation of dyads, formed when two units (e.g. CL–CL vs. CL–TMC) are next to each other in the backbone. Furthermore, the evaluation of the random character of the PCT samples was verified via13C-NMR. Fig. S8b† displays the carbonyl region (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) of both CL and TMC units, the evolution and extinction of peaks (e.g. TTT or CCC) while varying the copolymer composition has been attributed to the random character of the materials.
O) of both CL and TMC units, the evolution and extinction of peaks (e.g. TTT or CCC) while varying the copolymer composition has been attributed to the random character of the materials.
In Table S1† the values of the monomer conversion, practical copolymer composition, number average molecular weight (Mn) and polydispersity are given. According to the 1H-NMR analysis, the practical values of Mn correspond closely to the theoretical ones. Noticeably in this analysis, the CL conversion during the polymerization remained high (exceeding 99.3%) whereas TMC was slower to convert (lowest value at 88.4%), suggesting that the polymerization kinetics favor the CL monomer.
According to the gel permeation chromatography (GPC) traces (Fig. S29†) the PCT polymers presented dispersities (Đ) in the range of 1.20 to 1.54, in line with previous reports.26 Seen in the GPC, as the amount of TMC increases, the distribution of the curves tends to be broader. Moreover, the corresponding Mn values estimated by the GPC are higher than those obtained with the NMR analysis. This overestimation of the molecular weight distribution by GPC in TMC containing polymers has been previously reported.27 Transesterification, well-known to increase poydispersity in ring opening PCT polymerizations,28 is thought to also lead to this overestimation.
The synthesis of the LDI-HEA endcapping reagent is illustrated in Scheme S2† and was characterized by FTIR (ESI†). Of note, this reaction was run in a 2:1 molar HEA:LDI ratio, which will produce a mixture of mono (LDI-HEA), di (HEA-LDI-HEA), and unreacted (LDI). The obtained mixture of products was utilized without further purification.
The final printable macro-crosslinkers were prepared by reacting the PCT prepolymers with the LDI-HEA endcapping reagent at 40 °C in dry toluene for 24 h. The FTIR spectra of the products show the disappearance of the broad OH band at 3550 cm−1 and the appearance of signals at 3369 cm−1 and 1523 cm−1 corresponding to the N–H of the amide moiety from the attached LDI-HEA (ESI†). Shown in Fig. 1 with the 50:50 polymer as an example, the NMR spectra show the characteristic peaks of CL (4.06, 2.31, 1.65 and 1.39 ppm), TMC (4.24 and 2.05 ppm) and acrylate double bond (6.44, 6.15 and 5.96 ppm). All macro-crosslinkers synthesized showed a characteristic shift in GPC traces, attributed to chain extension via the LDI-HEA mixture (see ESI† for GPC traces).
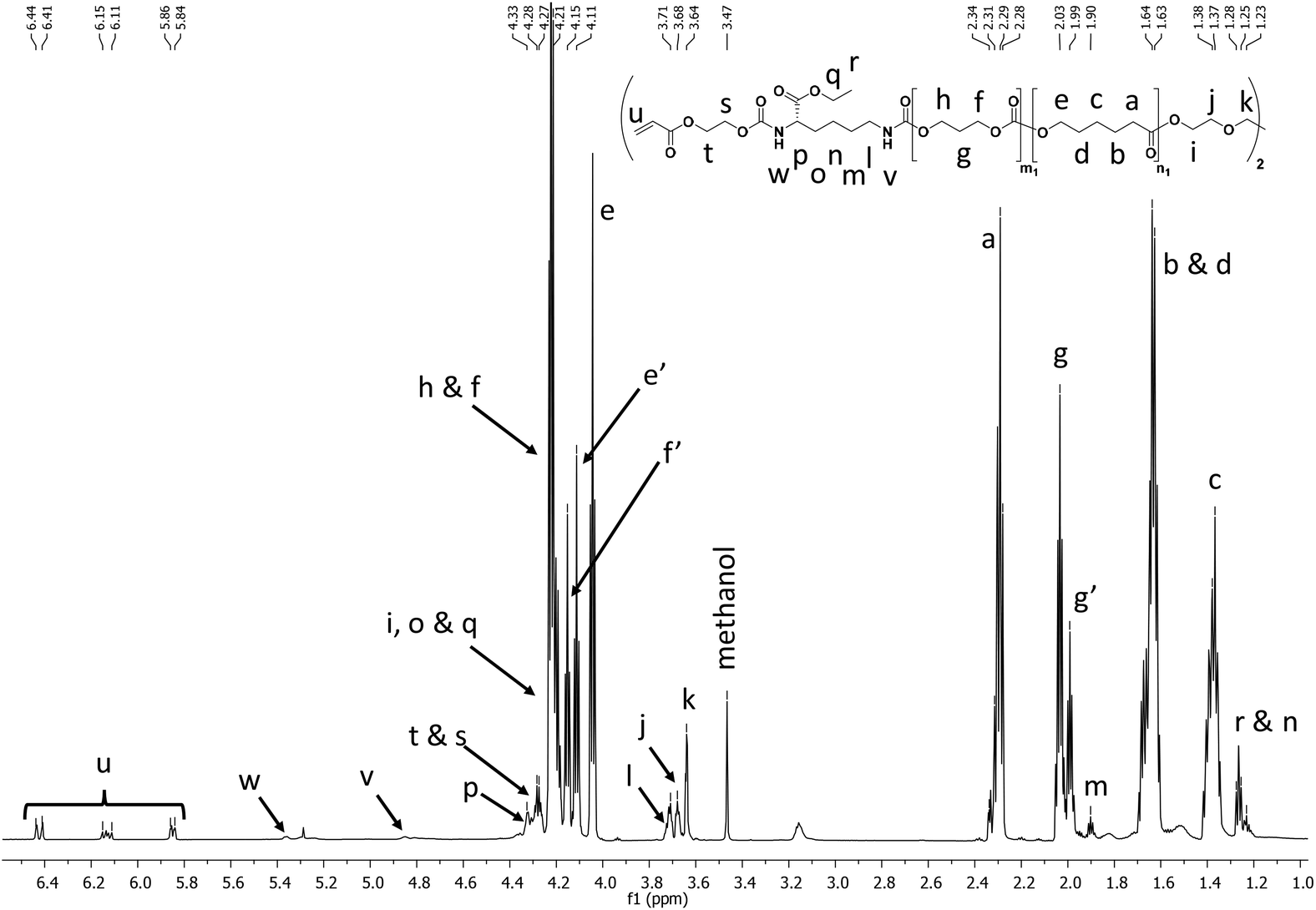 | ||
| Fig. 1
1H NMR in CDCl3 showing the characteristic peaks for CL, TMC, LDI, and acrylate protons in the final polymer. The PCTAc 50:50 copolymer is shown as a representative example. | ||
PCTAc films were prepared by pouring a pre-mixed resin (70 wt% PCTAc, 29 wt% reactive diluent, 1% TPO) into a glass Petri dish and curing it in a UV oven with a 365 nm wavelength for 5 min from top and bottom. All polymeric compositions formed free-standing films, which looked clear after post curing; however, samples with high CL (90 and 75%) content formed semi-crystalline polymers. Opacity in the printed samples were observed after 2 h (90:10) and 24 h (75:25) at room temperature.
DSC analysis of the different photopolymerized and crosslinked PCTAc films are shown in Fig. 2c. Films with a high content of PCL showed melting peaks at around 40 °C (90:10/ΔHf = 37.60 J g−1) and 32 °C (75:25/ΔHf = 29.85 J g−1). In addition, the 75:25 (CL:TMC) sample exhibited a cold crystallization peak at −17 °C, which is attributed to the larger amount of TMC disrupting PCL crystallization. This observation is in line with the slow crystallization kinetics observed in the printed constructs (vide supra). Polymers with more than 50% TMC remain amorphous. Another interesting observation is the shift of the glass transition temperature from −56 °C to −27 °C with an increase of TMC content.
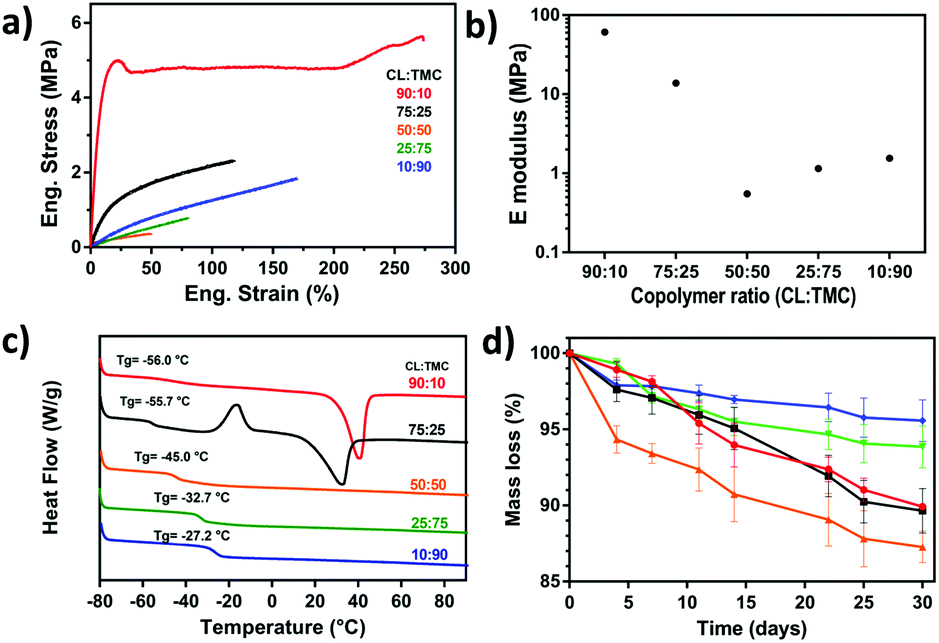 | ||
| Fig. 2 (a) Engineering stress–strain curves and, (b), Young's modulus of DLP fabricated films with 90:10, 75:25, 50:50, 25:75 and 10:90 CL:TMC ratios. (c) Dynamic scanning calorimetry (DSC) traces of PCTAc resins and (d) mass loss (%) over time during accelerated degradation studies. | ||
Determination of the tensile mechanical properties was performed after drying under reduced pressure. Fig. 2b illustrates the Young's modulus of the 5 different samples. The photocured PCTAc with higher content of CL monomer (90:10 and 75:25) displayed a stiff behavior, whereas the remaining samples were flexible materials. Interestingly, the 90:10 copolymer sample presented a different mechanical response compared to the rest of the samples. The 90:10 sample underwent a color transition from transparent to white after several hours at RT, thus it is hypothesized that the CL crystalline domains act as physical fillers, and thus enhancing the mechanical properties of this PCTAc.29 When the amount of amorphous TMC increased, samples became more brittle and showed lower strain at break. The sample with equimolar amounts of CL and TMC (50:50) presented the lowest mechanical properties. PTMC chains are known to undergo a strain induced crystallization, which can be observed in a slight increase of mechanical properties for the samples of 25:75 and 10:90 compared to 50:50.30 The large effect of crystallinity on storage modulus, coupled with the subtle effect of TMC, results in the trends across the series.
Table S2† shows the mechanical data for all PCTAc films as directly cured in a UV oven. Films with high CL concentrations showed the highest values of Young's modulus up to 61 MPa. Films with increasing TMC content showed lower Young's moduli as the amorphous phase increased and the Young's modulus dropped to values between 0.6 and 1.6 MPa. We did observe that the storage moduli of DLP printed films were slightly lower than directly cured films (Table S3†). This difference is currently hypothesized to arise from lower kinetic chain length in the DLP fabricated films. Nevertheless, the tensile moduli of these materials are comparable for soft tissues like human blood vessels (E = 1.5–3 MPa) or human myocardium (E = 0.2–0.5 MPa), currently an overlooked mechanical range for DLP resins.31
To investigate the potential of the PCTAc materials as tissue regeneration scaffolds, biocompatibility studies were also performed (Fig. 3). Human Mesenchymal Stromal Cells (hMSCs), known for their sensitivity to materials mechanical and chemical properties, were cultured on DLP fabricated PCTAc films. hMSCs readily attached after only 24 h of culture and showed differential morphologies that were dependent on the CL:TMC ratios (Fig. 3a). Cells cultured on copolymers with high CL:TMC ratios (90:10 and 75:25) appeared clustered and with small apparent surface spread area, while copolymers with 50:50, 25:75 and 10:90 CL:TMC ratios showed spindle- and pancake-shapes with increasing apparent cell spread areas and stress-fiber formation with decreased CL:TMC ratios. Moreover, higher cell densities were observed on substrates with lower CL:TMC ratios. In agreement with this observation, quantification of DNA on copolymer films showed a higher cell number, of approximately 12000 cells, attached to copolymers with 50:50, 25:75 and 10:90 as compared to the approximately 5000 cells attached to 75:25 and 90:10 CL:TMC ratios (Fig. 3b). These results suggest that the addition of TMC promotes a better cell adhesion as compared to traditional PCL homopolymers, known for their poor cell adhesive properties. Cell viability studies showed values of nearly 100% viable cells, similar to that of TCP controls (Fig. 3a and c). In accordance to the viability test, cells cultured on PCTAc films showed similar or lower lactate dehydrogenase (LDH) release values than the negative controls, supporting their high cytocompatibility (Fig. 3d). PCTAc films of 25:75 monomer ratio showed a slightly higher LDH release of 10%, which are comparable with other biocompatible resins and materials.32
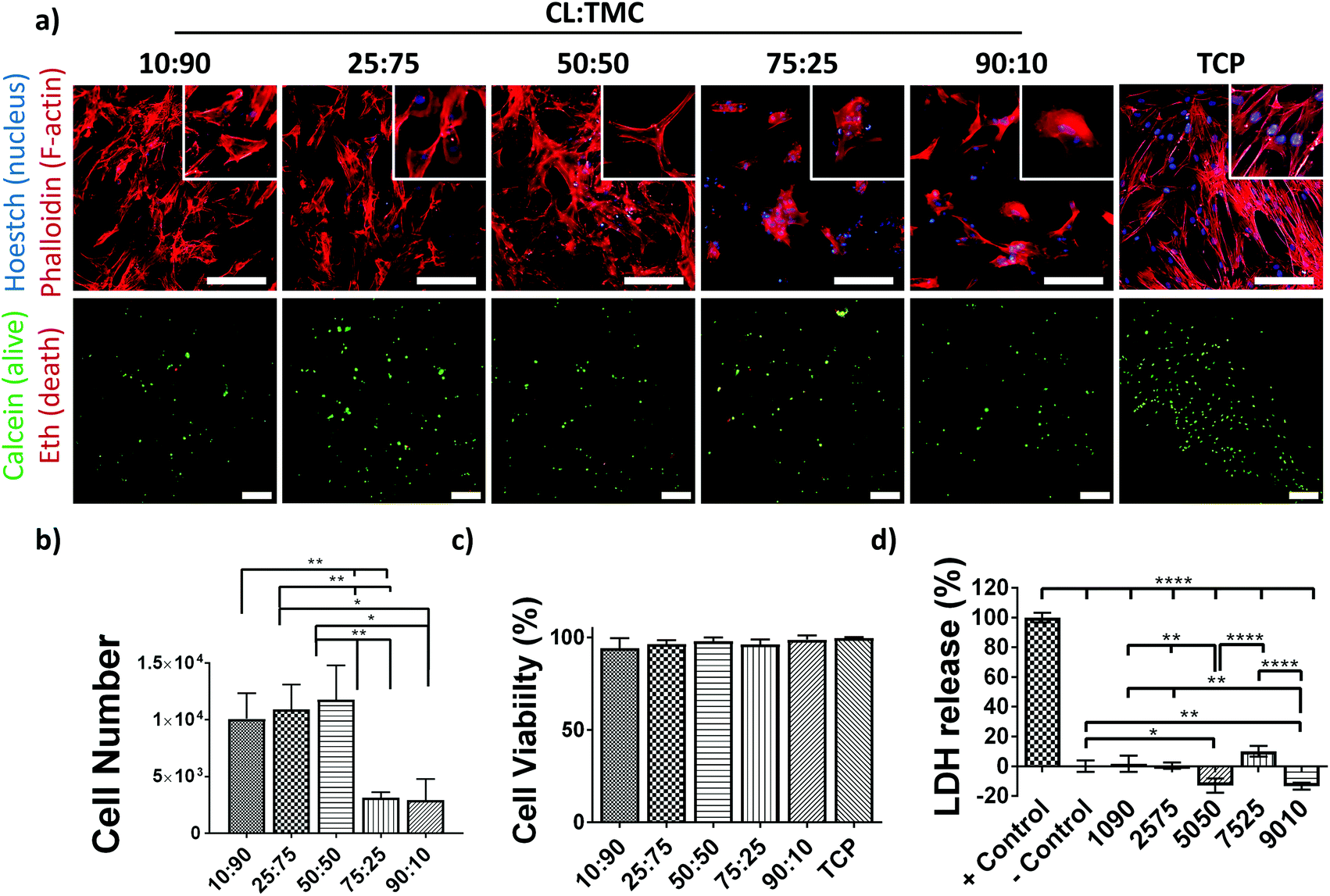 | ||
| Fig. 3 Fluorescence microscopy images of hMSCs cultured on DLP films consisting of various ratios of CL:TMC as well as tissue culture polystyrene plate (TCP) control. (a) To characterize cell attachment and morphology, hMSCs were stained after 24 h of culture with Hoestch (nucleus, blue) and phalloidin (F-actin, red) and, with calcein (green, nucleous, alive) and ethidium bromide homodimer-1 (Eth) (red, nucleus, dead) for viability experiments. Scale bar is 200 μm. (b) Cell number of attached cells to polymer films and TCP controls after 24 h of seeding. (c) Cell viability as quantified from images in (a). (d) Lactate dehydrogenase (LDH) release from hMSCs cultured on the films and of non-treated (+ control) and chemically lysed (− control) cells cultured on TCP. (d) Data is presented as mean ± SD, n = 3. | ||
These PCTAc materials were designed as biocompatible and bioresorbable formulations for soft tissue regeneration. Therefore, we performed an accelerated degradation test for 30 days under basic conditions (Fig. 2d). The polymeric resin with equimolar monomer composition (PCTAc 50:50) showed the fastest degradation, while a higher content of TMC slowed down the degradation rate. This effect has been already reported, and it is known that TMC is less sensitive to hydrolytic degradation than PCL.33 Furthermore, dimensional analysis of the materials during the degradation was conducted and results can be found in the ESI.† Ruling out any skew in the data due to swelling of the constructs, we also observed negligible (<2%) swelling of the films at 37 °C over 5 days (ESI†).
In order to evaluate the processability of PCTAc resins, 3D scaffolds of each resin type were prepared for printing by adding 2-(2-ethoxyethoxy)ethyl acrylate (EOEOEA) (30 wt%) and TPO (1 wt%). The sample of a printed structure is shown in Fig. 4.
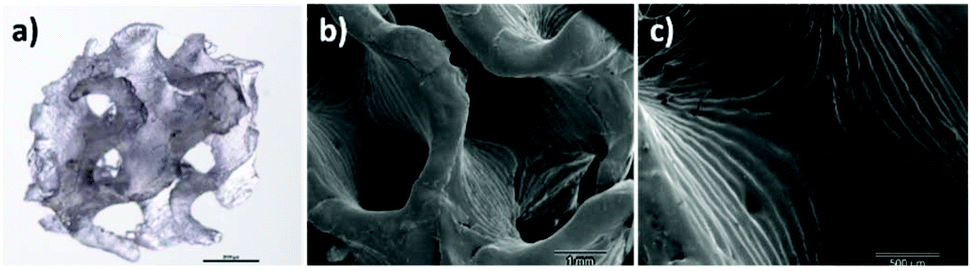 | ||
| Fig. 4 DLP fabricated 3D scaffold of PCTAc 25:75 resin with gyroid structure. (a) Optical microscopy image (scale bar 2 mm), (b) Scanning Electron Microscopy (SEM) image and (c) zoom-in showing the different printing layers. Scales are 1 mm and 500 μm, respectively. | ||
The synthesis of a well-defined co-polymer library has allowed us to explore a range of DLP polymerizable resins, with tunable mechanical properties and degradation rates, which show control over cell adhesion and spreading in fabricated scaffolds. By changing the monomer ratios of the polyester copolymer segment, the mechanical properties and the degradation rate are tunable and can be used to target specific tissues. All formulations are cytocompatible, and all formulation are capable of DLP scaffold fabrication into complex geometries. The polymers with a monomer ratio of 50:50 showed the lowest mechanical properties and the fastest degradation time.
These materials are straightforward to synthesize on large scales and are based on industrially translatable procedures. Through rational changes in the network and comonomer ratios, scaffolds with diverse degradation times and mechanical properties can be quickly fabricated into complex shapes using a single polymer platform. The versatility of this approach lends these polymers well to further in vitro and in vivo testing of DLP printed biodegradable scaffold for soft and medium-soft tissue regeneration. Future work will include the incorporation of functional handles and stimuli responsive elements within the polymer matrix.
Conflicts of interest
A. Dias (DSM Biomedical) is co-inventor on a patent (EP 2089438B, US 9458256) related to the creation of urethanes with acrylate functionalities.Acknowledgements
This work was supported financially by the Province of Limburg and the Brightlands Materials Center.Notes and references
- S. C. Ligon, R. Liska, J. Stampfl, M. Gurr and R. Mulhaupt, Chem. Rev., 2017, 117, 10212–10290 CrossRef CAS PubMed.
- K. Kowsari, B. Zhang, S. Panjwani, Z. Chen, H. Hingorani, S. Akbari, N. X. Fang and Q. Ge, Addit. Manuf., 2018, 24, 627–638 CrossRef CAS.
- F. P. Melchels, K. Bertoldi, R. Gabbrielli, A. H. Velders, J. Feijen and D. W. Grijpma, Biomaterials, 2010, 31, 6909–6916 CrossRef CAS PubMed.
- K. S. Lim, R. Levato, P. F. Costa, M. D. Castilho, C. R. Alcala-Orozco, K. M. A. van Dorenmalen, F. P. W. Melchels, D. Gawlitta, G. J. Hooper, J. Malda and T. B. F. Woodfield, Biofabrication, 2018, 10, 034101 CrossRef PubMed.
- S. Schuller-Ravoo, E. Zant, J. Feijen and D. W. Grijpma, Adv. Healthcare Mater., 2014, 3, 2004–2011 CrossRef PubMed.
- A. P. Zhang, X. Qu, P. Soman, K. C. Hribar, J. W. Lee, S. Chen and S. He, Adv. Mater., 2012, 24, 4266–4270 CrossRef CAS PubMed.
- K. Arcaute, B. Mann and R. Wicker, Acta Biomater., 2010, 6, 1047–1054 CrossRef CAS PubMed.
- N. A. Chartrain, C. B. Williams and A. R. Whittington, Acta Biomater., 2018, 74, 90–111 CrossRef CAS PubMed.
- R. Gautam, R. D. Singh, V. P. Sharma, R. Siddhartha, P. Chand and R. Kumar, J. Biomed. Mater. Res., Part B, 2012, 100, 1444–1450 CrossRef PubMed.
- D. A. Shimko and E. A. Nauman, J. Biomed. Mater. Res., Part B, 2007, 80, 360–369 CrossRef PubMed.
- J. A. Wilson, D. Luong, A. P. Kleinfehn, S. Sallam, C. Wesdemiotis and M. L. Becker, J. Am. Chem. Soc., 2018, 140, 277–284 CrossRef CAS PubMed.
- S. R. Petersen, J. A. Wilson and M. L. Becker, Macromolecules, 2018, 51, 6202–6208 CrossRef CAS.
- Y. Luo, C. K. Dolder, J. M. Walker, R. Mishra, D. Dean and M. L. Becker, Biomacromolecules, 2016, 17, 690–697 CrossRef CAS PubMed.
- S. Bertlein, G. Brown, K. S. Lim, T. Jungst, T. Boeck, T. Blunk, J. Tessmar, G. J. Hooper, T. B. F. Woodfield and J. Groll, Adv. Mater., 2017, 29, 1703404 CrossRef PubMed.
- L. Elomaa, S. Teixeira, R. Hakala, H. Korhonen, D. W. Grijpma and J. V. Seppala, Acta Biomater., 2011, 7, 3850–3856 CrossRef CAS PubMed.
- F. P. Melchels, J. Feijen and D. W. Grijpma, Biomaterials, 2009, 30, 3801–3809 CrossRef CAS PubMed.
- B. van Bochove, G. Hannink, P. Buma and D. W. Grijpma, Macromol. Biosci., 2016, 16, 1853–1863 CrossRef CAS PubMed.
- M. N. Cooke, J. P. Fisher, D. Dean, C. Rimnac and A. G. Mikos, J. Biomed. Mater. Res., Part B, 2003, 64, 65–69 CrossRef PubMed.
- J. J. Rongen, B. van Bochove, G. Hannink, D. W. Grijpma and P. Buma, J. Biomed. Mater. Res., Part A, 2016, 104, 2823–2832 CrossRef CAS PubMed.
- K. Fukushima, Biomater. Sci., 2016, 4, 9–24 RSC.
- T. Matsuda and M. Mizutani, J. Biomed. Mater. Res., 2002, 62, 395–403 CrossRef CAS PubMed.
- E. Bat, J. A. Plantinga, M. C. Harmsen, M. J. van Luyn, J. Feijen and D. W. Grijpma, J. Biomed. Mater. Res., Part A, 2010, 95, 940–949 CrossRef PubMed.
- L. Timbart, M. Y. Tse, S. Pang and B. G. Amsden, Materials, 2010, 3, 1156–1171 CrossRef CAS.
- I. H. Pereira, E. Ayres, P. S. Patricio, A. M. Goes, V. S. Gomide, E. P. Junior and R. L. Orefice, Acta Biomater., 2010, 6, 3056–3066 CrossRef CAS PubMed.
- J. Ling, W. Zhu and Z. Shen, Macromolecules, 2004, 37, 758–763 CrossRef CAS.
- I. Palard, M. Schappacher, B. Belloncle, A. Soum and S. M. Guillaume, Chemistry, 2007, 13, 1511–1521 CrossRef CAS PubMed.
- P. Dobrzynski, M. Pastusiak and M. Bero, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 1913–1922 CrossRef CAS.
- A. Schindler, Y. M. Hibionada and C. G. Pitt, J. Polym. Sci., Polym. Chem. Ed., 1982, 20, 319–326 CrossRef CAS.
- S. Wang, M. J. Yaszemski, J. A. Gruetzmacher and L. Lu, Polymer, 2008, 49, 5692–5699 CrossRef CAS PubMed.
- A. P. Pêgo, D. W. Grijpma and J. Feijen, Polymer, 2003, 44, 6495–6504 CrossRef.
- H. Jawad, N. N. Ali, A. R. Lyon, Q. Z. Chen, S. E. Harding and A. R. Boccaccini, J. Tissue Eng. Regener. Med., 2007, 1, 327–342 CrossRef CAS PubMed.
- M. C. Serrano, R. Pagani, J. Pena and M. T. Portoles, Biomaterials, 2005, 26, 5827–5834 CrossRef CAS PubMed.
- J. Yang, F. Liu, L. Yang and S. Li, Eur. Polym. J., 2010, 46, 783–791 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials, methods and figures. See DOI: 10.1039/c9bm01042d |
| ‡ Authors share co-authorship. |
| This journal is © The Royal Society of Chemistry 2019 |