
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient blue light emitting materials based on m-carborane–anthracene dyads. Structure, photophysics and bioimaging studies†
Mahdi
Chaari
 a,
Zsolt
Kelemen
a,
Duane
Choquesillo-Lazarte
b,
Nerea
Gaztelumendi
c,
Francesc
Teixidor
a,
Clara
Viñas
a,
Carme
Nogués
*c and
Rosario
Núñez
*a
a,
Zsolt
Kelemen
a,
Duane
Choquesillo-Lazarte
b,
Nerea
Gaztelumendi
c,
Francesc
Teixidor
a,
Clara
Viñas
a,
Carme
Nogués
*c and
Rosario
Núñez
*a
aInstitut de Ciència de Materials de Barcelona (ICMAB-CSIC), Campus U.A.B., 08193, Bellaterra, Barcelona, Spain. E-mail: rosario@icmab.es; Fax: +34 93 580 5729; Tel: +34 93 580 1853
bLaboratorio de Estudios Cristalográficos, IACT-CSIC, Avda. de las Palmeras 4, 18100 – Armilla, Granada, Spain
cDepartament de Biologia Cellular, Fisiologia i Immunologia. Universitat Autònoma de Barcelona, Campus U.A.B., 08193, Bellaterra, Barcelona, Spain. E-mail: Carme.Nogues@uab.cat
First published on 2nd October 2019
Abstract
Efficient monosubstitution of the non-iodinated, mono-iodinated and di-iodinated m-carborane cluster at one Ccluster has led to the preparation of three single organic molecule-carborane dyads (4–6), which exhibited exceptional fluorescence properties with quantum yield values of 100% in solution, for all of them, with maxima around 415 nm, which correspond to the locally excited state (LE) emission. These results suggest that simply linking the m-carborane fragment to one anthracene unit through a CH2 spacer produces a significant enhancement of the fluorescence in the final fluorophore, probably due to the free rotation of the anthracene linked to the Ccluster. Besides, the presence of one or two iodine atoms linked to boron atoms does not cause any influence on the photophysical properties of the dyads, as it is confirmed by TD-DFT calculations. Notably, the three conjugates show good fluorescence efficiency in the aggregate state with quantum yields in the range of 19–23%, which could be ascribed to the presence of CH2, particularly for 4, and the iodine atoms in 5 and 6, which prevent π–π stacking. All these results indicate that our dyads are extremely good emitters in solution while maintaining the emission properties in the aggregate state. Crystal packing, fingerprint plot analysis, and TD-DFT calculations for the three compounds support these results. Confocal microscopy studies show that 6 is the best-internalized compound by HeLa cells via endocytosis, although 4 and 5 also presented a high fluorescence intensity emission. Moreover, due to the blue emission, this compound is an excellent candidate to be applied as a fluorescent dye in bioimaging studies.
Introduction
Anthracene is a π-conjugated system widely investigated in both chemistry and materials science, due to its significant photophysical, photochemical and chemical properties.1 Owing to their excellent luminescence properties, anthracene derivatives have potential applications in organic functional materials, such as organic field effect transistors (OFETs) and organic light-emitting diodes (OLEDs), and for sensing metal ions, pH, small organic molecules, etc.2,3 On the other hand, the excimer emission from anthracene is a well reported phenomenon, which is of special interest in photochemistry, supramolecular chemistry, photobiology and polymer science.4 The excimer formation of anthracene is from its excited dimer, which has four possible structures, such as normal dimer, twisted dimer, offset dimer and T-shaped dimer.5 Usually, planar aromatic molecules can easily form an excimer in concentrated solutions or in the solid state because of the intermolecular π–π interactions, which play a crucial role in excimer formation. Usually, the excimer exhibits red-shifted, broadened and structureless fluorescence emission with regard to that of its monomer.Carborane clusters are fascinating chemical species that have been the subject of intense and attractive research over the last six decades.6 They are very robust compounds characterized by a unique three-dimensional (3D) σ-delocalization,7 high thermal and chemical stability,8 hydrophobicity, metabolic inertness and low toxicity in biological systems.6d–f,9 Carboranes show high versatility toward functionalization, which makes them suitable building blocks easily linkable to molecular and polymeric π-conjugated systems.10 One of the main features of the o-carborane cluster is its electron-accepting ability through substitution at the Ccluster (Cc),11 when it is linked to an aryl donor group, and subsequently accepts an excited charge via intramolecular charge transfer (ICT) from the donor group. These ICT states are influenced by the Cc–Cc bond vibration,12 which can vibrate and relax non-radiatively or radiatively giving a red-shift emission.13,14 Moreover, these systems that show low or null fluorescence efficiency in solution usually exhibit aggregation-induced emission (AIE) in the solid state, due to the restriction of the molecular motion, which leads to a significant increase in the emission quantum yield.15
Over the last years, we have designed carborane-containing single molecules, whose emission in solution depends critically on the cluster isomerism (o- or m-) and the fluorescence efficiency can be tailored by changing the substituent at the adjacent Cc.8,16 It was observed that molecules showing moderate emission in solution were able to exhibit good fluorescence efficiency in the solid state.17 In this way, and depending on the targeted application, we are capable of preparing highly fluorescent materials in both states. Noticeably, m-carborane has been demonstrated to be a perfect platform to boost the photoluminescence properties of organic π-conjugated systems linked to it, both in solution and the solid state.16,17
Some examples of anthracene-o-carborane dyads and triads where the anthracene is directly linked to the Cc of the o-carborane or through a π-conjugated spacer between both fragments have been reported.18 It is well described that o-carborane acts as a strong electron-withdrawing unit in the excited state of these conjugated systems causing a rapid ICT from the anthracene to the o-carborane, with a subsequent quenching of the locally excited state (LE) fluorescence emission in solution. Moreover, these dyads have shown highly-efficient solid-state emission, as well as mechanochromic and thermochromic luminescence properties.18 Noticeably, m-carborane–anthracene triads obtained by linking two anthracene units to the m-carborane fragment exhibited a significant increase in more than two-fold in the intrinsic fluorescence quantum yield of the anthracene in solution as well as a red-shift of the emission maximum and moderate quantum efficiencies in the aggregate state.19 Nevertheless, the differences in the ϕF values obtained for different triads in the aggregate state were attributed to the arrangement of dimers in the solid state structures; it was concluded that the presence of a large number of BH⋯I contacts leads to a less delocalized system for the diiodo derivative, and therefore a lower quantum yield value.
We present here a set of three m-carborane–anthracene dyads efficiently synthesized by Cc monosubstitution of the m-carborane with one anthracene group through a CH2 unit. Iodination of one and two B atoms is also performed to produce B–I vertices. These systems were designed to enhance the photoluminescence (PL) properties with regard to previously reported m-carborane–anthracene triads, both in solution and the solid state. The crystal structures of the synthesized compounds have been established by X-ray diffraction analysis. Photophysical analysis in solution and the aggregate state have been performed and complemented with TD-DFT theoretical calculations to establish a meaningful structure–photophysical property relationship. A comparison between the current dyads and related triads is also reported. Owing to our interest in the biological applications of fluorescent carborane derivatives, the cytotoxicity and internalization of the dyads and their cellular localization in HeLa cancer cells are also discussed.
Results and discussion
Synthesis and characterization of anthracenyl-monosubstituted m-carborane derivatives 4–6
The synthesis of compounds 4–6 was achieved by nucleophilic substitution at one Cc atoms,20 following a similar procedure to that used for the previous o- and m-carborane derivatives.16,17,19 Compound 4 was obtained by the nucleophilic substitution of 1,7-closo-C2B10H12 (1), whereas 5 and 6 were obtained from the respective mono- and di-iodinated derivatives, 9-I-1,7-closo-C2B10H11 (2) and 9,10-I2-1,7-closo-C2B10H10 (3), which were previously prepared by electrophilic substitution at the B atoms.21 The key step for the preparation of compounds is the monolithiation of starting compounds 1–3, which was efficiently performed using THF as a solvent under diluted conditions at 0 °C; then the reaction of the corresponding monolithium salts with one equiv. of 9-chloromethyl anthracene at reflux overnight gave compounds 4, 5 and 6, respectively, in 52, 54 and 41% yields, respectively (Scheme 1). The nucleophilic substitution reactions were monitored by 11B{1H} NMR following the appreciable changes in the boron resonance distributions. Although for m-carborane the monosubstitution at the Ccluster is relatively easier than for o-carborane, in both cases a mixture of mono- and di-substituted species is always obtained, even on controlling the reaction conditions. Proof of this is that only a few examples of mono-substituted derivatives from m-carborane have been reported in the literature.22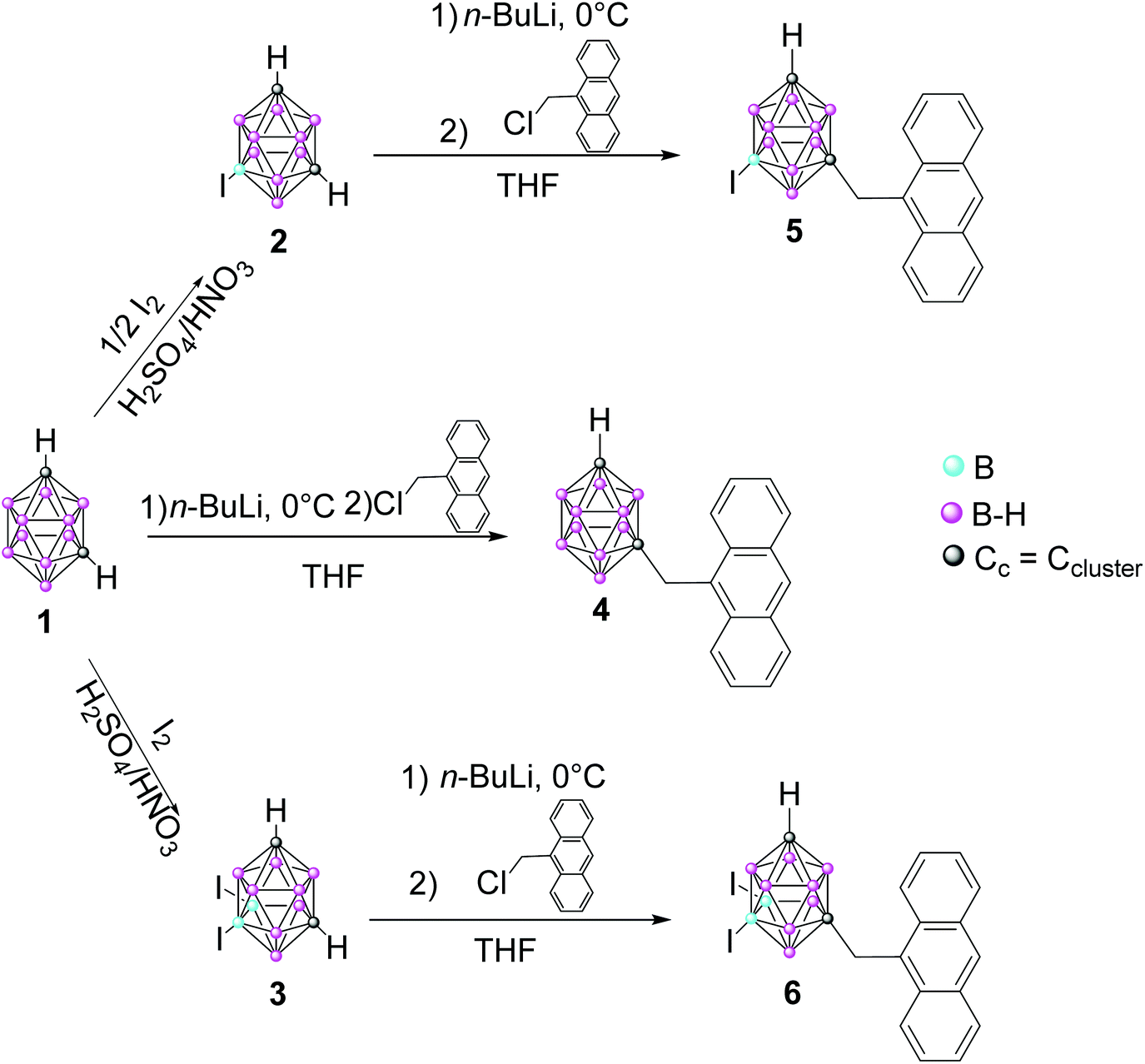 | ||
| Scheme 1 Procedure to obtain compounds 4–6. | ||
The structures of 4–6 were established on the basis of IR, 1H, 13C{1H} and 11B{1H} spectroscopy and elemental analysis and all of them were confirmed by X-ray diffraction analysis. On the one hand, IR spectra show typical ν(B–H) strong bands for closo clusters between 2556 and 2609 cm−1. On the other hand, 1H NMR spectra of 4–6 display one new singlet at around δ = 4.30 ppm due to the Cc–CH2 protons. Then, compounds 4–6 show a Cc–H single resonance in the region at 2.75–2.92 ppm, which is slightly shifted upfield (approximately 0.2 ppm) with regard to starting compounds 1–3. Due to the mono and diiodination of the m-carborane cluster, the 11B{1H} NMR spectra for the three compounds are different; m-carborane derivative 4 shows resonances in the typical closo region, from δ −3.66 to −15.58 ppm, with the general pattern 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:2:2; mono-iodinated compound 5 shows resonances in the range from δ −2.65 to −23.87 ppm, with the 1:4:1:1:1:1:1 pattern, whereas di-iodinated compound 6 shows resonances in the range from δ −1.62 to −21.15 ppm, with the 1:1:2:2:2:2 pattern. Moreover, it is remarkable that the two B–H resonances at high frequency for all the anthracene derivatives 4–6 were shifted to downfield at around 1.5 ppm and the ranges between them are larger with regard to 1–3 (see the ESI†). As for the previously reported iodinated derivatives,23 the B–I are indubitably identified as the highest field resonances in the range from δ −21.15 to −23.87 ppm, which remain as a singlet in the 11B NMR. The 13C{1H} NMR spectra of 4–6 show one resonance at around δ = 55.10 ppm that is assigned to the Cc–CH2 carbons, which is shifted downfield (approximately 1 to 1.6 ppm) in 5 and 6 compared to 4. In contrast, one resonance at around δ = 33.88 ppm, which is slightly shifted upfield in 5 and 6 compared to 4, is assigned to the Cc–H carbons. All compounds show aromatic resonances in the range from δ 124.17 to 131.53 ppm. Elemental analyses also confirmed the stoichiometry of all these compounds (see the Experimental section).
:4:2:2; mono-iodinated compound 5 shows resonances in the range from δ −2.65 to −23.87 ppm, with the 1:4:1:1:1:1:1 pattern, whereas di-iodinated compound 6 shows resonances in the range from δ −1.62 to −21.15 ppm, with the 1:1:2:2:2:2 pattern. Moreover, it is remarkable that the two B–H resonances at high frequency for all the anthracene derivatives 4–6 were shifted to downfield at around 1.5 ppm and the ranges between them are larger with regard to 1–3 (see the ESI†). As for the previously reported iodinated derivatives,23 the B–I are indubitably identified as the highest field resonances in the range from δ −21.15 to −23.87 ppm, which remain as a singlet in the 11B NMR. The 13C{1H} NMR spectra of 4–6 show one resonance at around δ = 55.10 ppm that is assigned to the Cc–CH2 carbons, which is shifted downfield (approximately 1 to 1.6 ppm) in 5 and 6 compared to 4. In contrast, one resonance at around δ = 33.88 ppm, which is slightly shifted upfield in 5 and 6 compared to 4, is assigned to the Cc–H carbons. All compounds show aromatic resonances in the range from δ 124.17 to 131.53 ppm. Elemental analyses also confirmed the stoichiometry of all these compounds (see the Experimental section).
X-ray structural analysis
Single crystals suitable for X-ray structural determination of compounds 4, 5 and 6 were obtained by slow evaporation from a mixture of chloroform/n-heptane (9:1), chloroform or dichloromethane at room temperature, respectively. The molecular structures for 4–6 were established by single crystal X-ray diffraction (Fig. 1) and are in agreement with the NMR data (vide supra). Experimental crystal data and structure refinement parameters for all structures reported in this work are listed in Table 1. Whereas compound 4 crystallizes in the triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group, compound 5 crystallizes in the monoclinic P21/n space group and compound 6 crystallizes in the orthorhombic Ima2 space group. The molecular structures for all these compounds show typical icosahedral geometry with very similar bond distances and angles, which are also similar to those in anthracenyl-disubstituted m-carborane compounds.19
space group, compound 5 crystallizes in the monoclinic P21/n space group and compound 6 crystallizes in the orthorhombic Ima2 space group. The molecular structures for all these compounds show typical icosahedral geometry with very similar bond distances and angles, which are also similar to those in anthracenyl-disubstituted m-carborane compounds.19
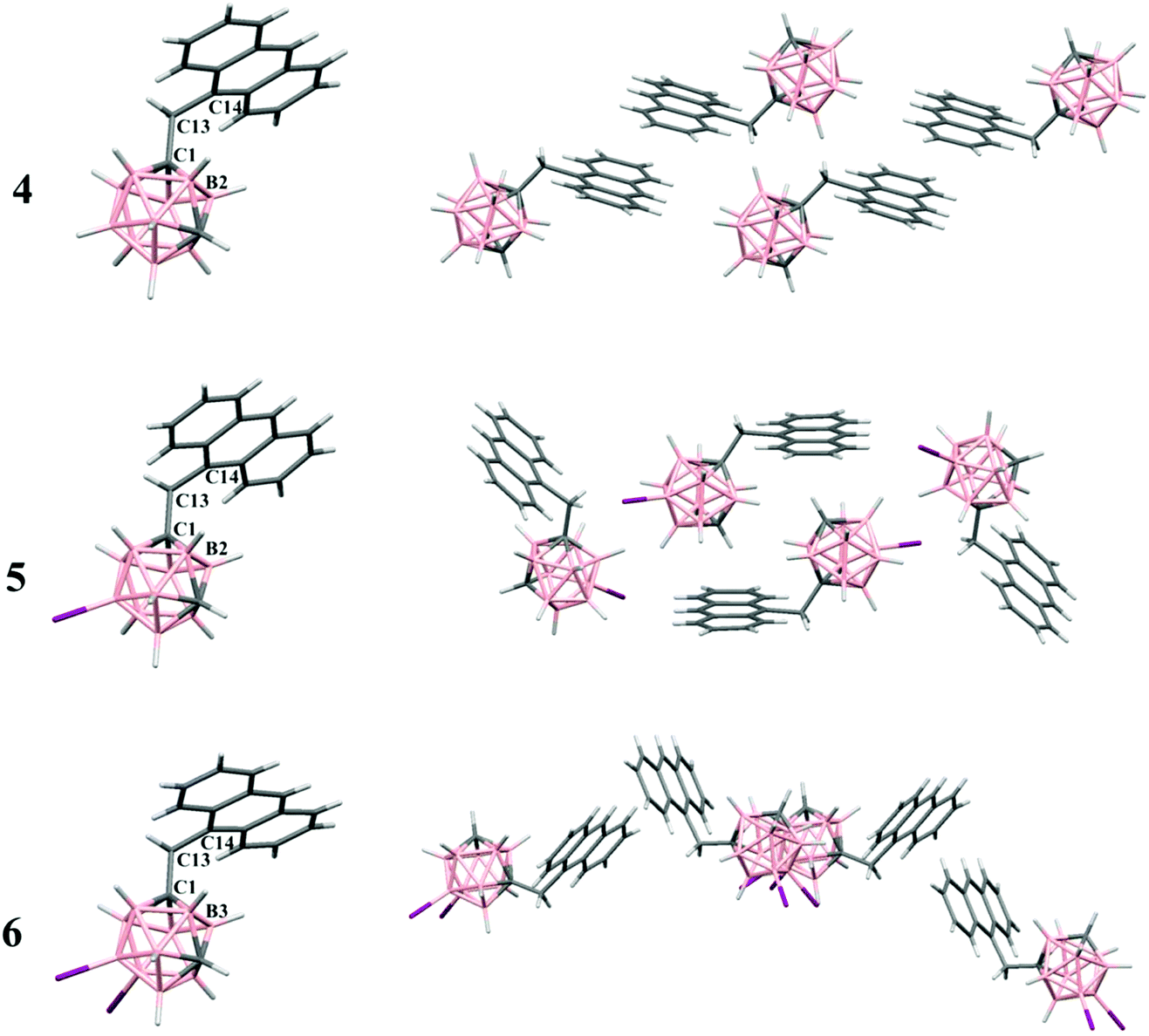 | ||
| Fig. 1 (Left column) Molecular structures of 4, 5 and 6. Torsion angles: 4 (C14C13C1B2, 37.7(2)°), 5 (C14C13C1B2, 41.1(4)°), and 6 (C14C13C1B3, 29°); (right column) projections showing the organization of four molecules of the compounds in the solid state. Color code: B pink; C grey; H white; and I violet. | ||
| 4 | 5 | 6 | |
|---|---|---|---|
| CCDC 1907569 (4), 1907570 (5) and 1907571 (6)† contain the supplementary crystallographic data for this paper. | |||
| Empirical formula | C17H22B10 | C17H21B10I | C17H20B10I2 |
| Formula weight | 334.44 | 460.34 | 586.23 |
| Crystal system | Triclinic | Monoclinic | Orthorhombic |
| Space group |
P |
P21/n | Ima2 |
| Temperature/K | 293 | 293 | 298 |
| Wavelength/Å | 0.71073 | 0.71073 | 0.71073 |
| a/Å | 6.9712(5) | 9.3190(8) | 13.801(3) |
| b/Å | 11.7604(7) | 11.7553(6) | 25.643(5) |
| c/Å | 12.9797(6) | 18.5808(12) | 6.8626(13) |
| α/° | 107.511(2) | 90 | 90 |
| β/° | 102.996(3) | 94.913 | 90 |
| γ/° | 102.125(3) | 90 | 90 |
| Volume/Å3 | 943.63(10) | 2028.0(2) | 2428.7(8) |
| Z | 2 | 4 | 4 |
| Density (calculated)/Mg m−3 | 1.177 | 1.508 | 1.603 |
| F(000) | 348 | 904 | 1112 |
| Theta range for data collection/° | 1.903 to 27.483 | 2.052 to 27.495 | 2.803 to 25.014 |
| Absorption coefficient/mm−1 | 0.058 | 1.579 | 2.591 |
| Goodness-of-fit on F2 | 1.030 | 1.123 | 1.038 |
| R 1 [I > 2sigma(I)] | 0.0577 | 0.0459 | 0.0405 |
| wR2 [I > 2sigma(I)] | 0.1464 | 0.0991 | 0.0766 |
| R 1 (all data) | 0.0874 | 0.0634 | 0.0603 |
| wR2 (all data) | 0.1624 | 0.1073 | 0.0791 |
In all structures, the m-carborane moiety is linked to one anthracene unit through a methylene spacer (–CH2–). As shown in Fig. 1, the rotation of the anthracene rings through the (H)(H)C–C(anthracene) bond allows more or less similar conformations in the solid state. Conformations found in the solid state and torsion angles are given in Fig. 1 caption.
The solid-state structures in 4–6 are mainly dominated by intermolecular C–H⋯anthracenyl (A) interactions (Fig. S1† and Table 2). Thus, extensive Cc–H⋯A interactions are found in all molecules, mainly between the Cc–H and B–H hydrogen atoms in 4 and 6, and C–H⋯π in 5 (Fig. S1† and Table 2). Moreover, interactions between –CH2– hydrogen atoms and aromatic Canthracene atoms are found in 4. Other weak B–H⋯I–B contact in 6 is listed in Table 2. From the crystal packing of 4 (Fig. 1 and Fig. S1†), we can see that there are no π–π stacking between two anthracenes, because the presence of the CH2 spacer, from which one hydrogen atom interacts with an aromatic Canthracene (2.82 Å), avoids the π–π packing from intermolecular anthracene groups that could allow the excimer formation or even a quenching of the fluorescence. In compounds 5 and 6, the iodine atoms dominate the crystal packing (Fig. 1 and Fig. S1†), obviously preventing π–π stacking between anthracene rings.
| Compounds | D–H⋯A | d(H⋯A) | ∠(DHA) | ∠(HHB) |
|---|---|---|---|---|
| Symmetry codes (i) 1 + x, y, z (ii) 1 − x, 1 − y, 2 − z (iii) 1 − x, 1 − y, 1 − z (iv) 0.5 − x, −0.5 + y, 0.5 − z (v) x, y, 1 + z (vi) 1 − x, 1 − y, z. | ||||
| 4 | C(13)–H(13B)⋯C(18)i | 2.828 | 122.5 | — |
| C(13)–H(13B)⋯C(19)i | 2.910 | 139.3 | — | |
| C(7)–H(7)⋯H(3)–B(3)ii | 2.484 | 120.5 | 166.6 | |
| 5 | C(7)–H(7)⋯Miii | 2.669 | 150.3 | — |
| C(17)–H(17)⋯H(11)–B(11)iv | 2.307 | 131.8 | 153.1 | |
| 6 | C(7)–H(7)⋯H(5)–B(5)v | 2.364 | 119.9 | 118 |
| C(7)–H(7)⋯I(1)–B(9)v | 3.103 | 135.3 | — | |
| B(4)–H(4)⋯I(1)–B(9)vi | 3.215 | 171.3 | — | |
From the fingerprint plot analysis, it can be found that H⋯H contacts comprise nearly 78% of the total Hirshfeld surface area for the non-iodinated compound 4 and it decreases to 65% and 52% for the iodinated compounds 5 and 6, respectively. The H⋯C contacts contribute around 13–19% to the total Hirshfeld surface area for the three compounds. The contribution of H⋯I interactions varies from 15% to 28% for 5 to 6, respectively (see more details in Fig. S2 in the ESI†).
DFT-calculations were carried out (see more details in the ESI†) and different rotamers were investigated for 4–6 (Fig. S3 in the ESI†). The structure of the most stable rotamer is identical to the rotamer obtained from the crystal structures. It should be noted that the energy differences between the rotamers are tiny and probably the rotational barrier is small, thus in solution there is not preferred orientation.
Photophysical properties and TD-DFT calculations
The photophysical properties of compounds 4–6 were determined by UV-Vis absorption and fluorescence spectroscopy in THF solutions (Table 3). Electronic properties of 4–6 in the ground state were assessed by UV-Vis absorption measurements (∼10−5 M, Fig. 2). All compounds exhibit similar absorption spectra showing vibrational structures assigned to the π–π* transition band of the anthracene moiety, with a sharp band around 258 nm and four peaks at around 334, 350, 369 and 389 nm. The molar absorption coefficients (ε) are slightly different for the three compounds, being in the range from 102 × 102 to 112 × 102 M−1 cm−1, where a slight increase in the value goes in parallel with the number of iodine atoms in the molecules.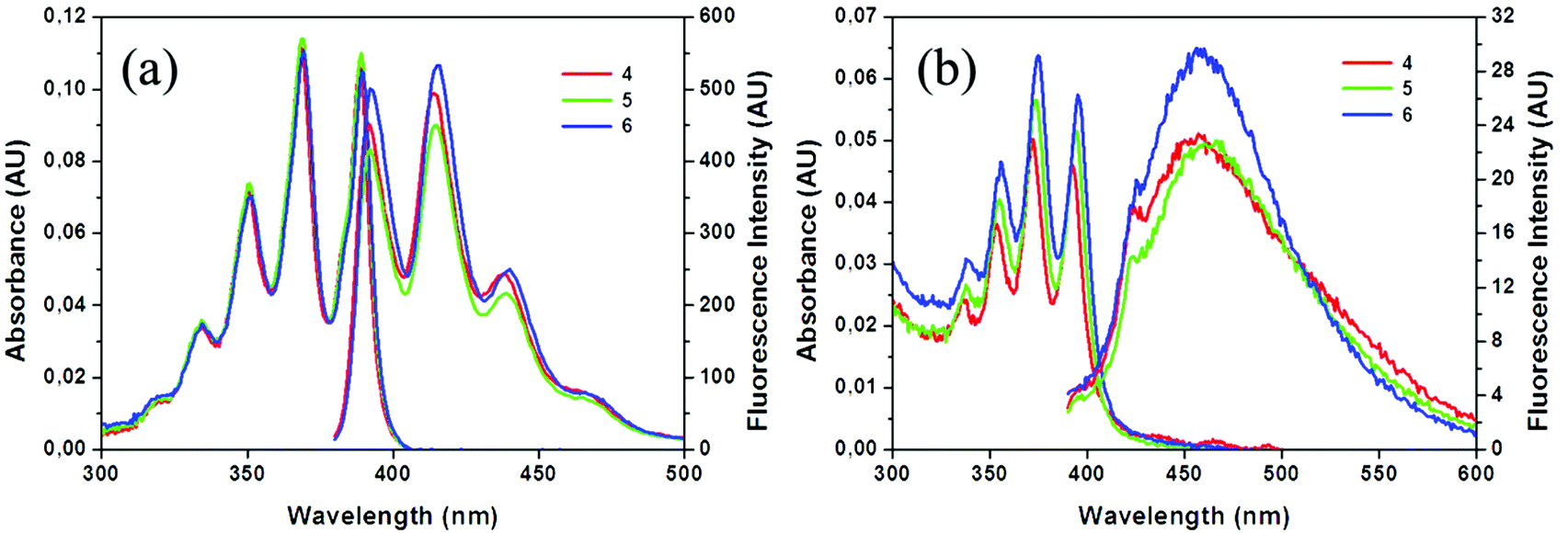 | ||
| Fig. 2 Absorption and emission spectra of 4 (red), 5 (green) and 6 (blue) in THF solutions (a) and aggregates (THF/H2O, v/v = 1/99) (b). AU: arbitrary units. | ||
| THF | THF/water (1/99) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compounds | λ abs (nm) | ε/105 (M−1 cm−1) | λ em (nm) |
ϕ
Fa |
Brightness 104 (M−1 cm−1) | Stokes shift (nm) | λ abs (nm) | λ em (nm) |
ϕ
Fa |
Stokes shift (nm) |
| a Reference compound quinine sulfate (0.5 M H2SO4, ϕF = 0.54). | ||||||||||
| 4 | 369 | 0.102 | 414 | 1 | 1.02 | 45 | 372 | 457 | 0.23 | 85 |
| 5 | 369 | 0.106 | 415 | 0.95 | 1.01 | 46 | 373 | 459 | 0.19 | 86 |
| 6 | 369 | 0.112 | 415 | 1 | 1.12 | 46 | 375 | 456 | 0.2 | 81 |
Fluorescence emission spectra of 4–6 were also measured in THF as well as in the aggregate state (THF/H2O = 1/99 (v/v), 1 × 10−5 M for 4–6). All of them show similar vibronic emission spectra in solution, with maxima at around λem = 415 nm (Fig. 2a), which can be assigned to the LE emission of the anthracene moiety. As for the previous compounds reported by us,19 the similarity between the spectra of 4–6 in solution and that of anthracene (λem = 420 nm) suggests that small electronic interactions between the anthracene units take place, and there is no influence of the iodine atom. Remarkably, compounds 4–6 exhibit extraordinary high fluorescence quantum yield values (ϕF) in solution that are around 100%. Table 3 summarises the photophysical data for all the compounds. It is worth noting that monosubstituted anthracene derivatives 4–6 exhibit significant higher fluorescence quantum yields in solution than the previously reported disubstituted m-carborane that contains two anthracene units, which exhibited ϕF values in the range of 63–66%.19
To gain more insight into the photophysical behaviour of 4–6 the Kohn–Sham orbitals and TD-DFT spectra were calculated at the B3LYP/6-31G* (for the iodine atom LANL2DZ basis set) level of theory. The B3LYP functional with 6-31G* basis set was successfully applied earlier for the investigation of related carborane based fluorophores.19,24 As it was noted the energy difference between the different rotamers is small and presumably all of them occur in solution; thus the frontier orbitals and TD-DFT were calculated for all rotamers (Fig. S3 in the ESI†). The orientation of the anthracene unit does not have a significant effect on the shape and the energy of the frontier orbitals, and as a consequence on the calculated transitions in the TD-DFT calculations (Fig. 3); the HOMO and LUMO are localized at the anthracene unit in all the investigated rotamers of 4–6 and TD-DFT calculations confirm the π–π* (HOMO–LUMO) transitions for 4–6. The energy of this transition exhibits an excellent numerical agreement with the observed spectra (Table S1 in the ESI†). Simple TD-DFT calculations could not describe the vibrational fine structure, which is specific for a rigid aromatic system such as anthracene. The calculated excitation refers to the transition from the ground state to the lowest vibrational level of the excited state, which corresponds to the band around 390 nm in the experimental spectra (calculated 389 nm). After investigating the influence of the iodine atoms, it could be established that they have no impact on the shape and the energy of the frontier orbitals, so they do not change the HOMO–LUMO transitions. On the other hand, the lone pairs of the iodine atoms do participate in the HOMO−1 and HOMO−2 for 5 and HOMO−X (X = 1–4) for 6. TD-DFT calculations demonstrate that the transitions from the lone pairs of the iodine atom(s) to the π* orbitals of the anthracene unit exist, but the probability of these transitions is low (the calculated oscillator strength is below 0.001); thus they could be not observed in the experimental spectra.
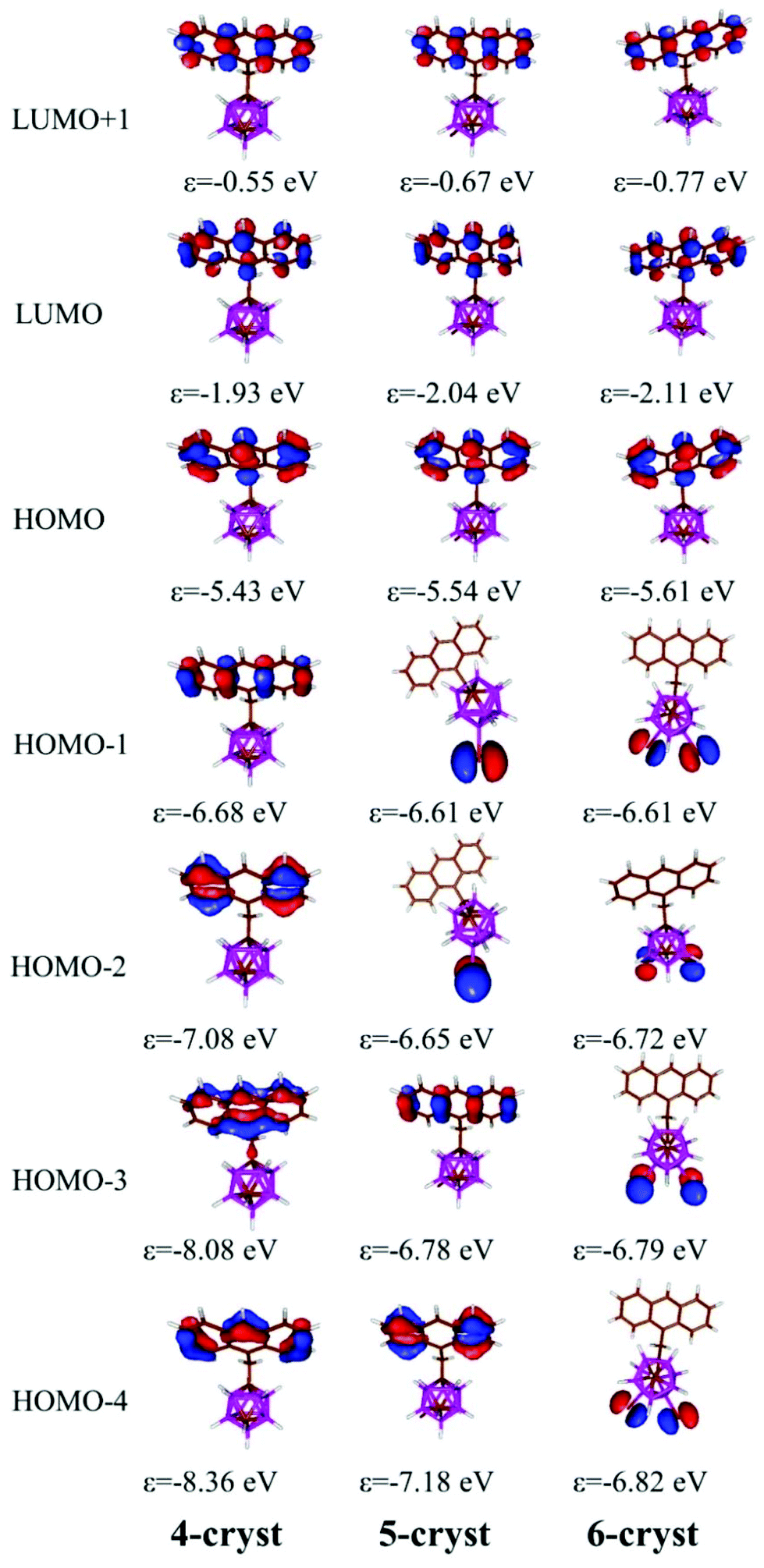 | ||
| Fig. 3 Some important Kohn–Sham molecular orbitals of 4-cryst, 5-cryst and 6-cryst and their energies in eV and at B3LYP/6-31G* level of theory. The order and the energy level of the orbitals are not strongly dependent on the orientation of the anthracene unit (different rotamers). | ||
Finally, we investigated the PL behaviour in the aggregate state (THF/H2O = 1/99 (v/v)); emission spectra of 4–6 are shown in Fig. 2b. The PL spectra for these compounds are very similar, showing non-vibronic structures and a maximum emission at around 456–459 nm (Table 3), which are about 40 nm red-shifted with respect to the THF solutions. Consequently, larger Stokes shifts are also observed. In general, all compounds were fluorescent in the aggregate state with ϕF values in the range of 19–23%. Nevertheless, a decrease in the quantum efficiency compared to the respective ones in solution (95–100%) is clearly observed, while maintaining the fluorescence properties. Investigating the literature data, similar emission spectra (with non-vibronic structures and maximum at around 450–460 nm) have been observed for anthracene excimers.4c,d,25 Our DFT calculations (see more details in the ESI, Fig. S4†) do not rule out the possible formation of excimers, but at this point it is difficult to establish a relationship between the emission spectra and the structure of our compounds, as all of them show similar emission properties although they have very different packing in the solid state.
Biological assays
Fluorescence imaging techniques have been used to visualize bio-components and bio-processes by transforming the chemical and biological information into detectable signals. Recently, we have reported that carborane–BODIPY dyads were easily internalized by HeLa cells, those bearing m-carborane clusters being the ones that show the best fluorescence characteristics for cell imaging purposes.26 Owing to our interest to use fluorescent carborane derivatives for biological purposes, herein we have analysed the cytotoxicity and the potential use of compounds 4–6 as fluorescent probes for biological systems.Cytotoxicity was analyzed by the Alamar Blue assay after HeLa cell incubation with each compound at 10 μM for 24 h. Only viable cells can reduce resazurin (the active compound of Alamar Blue) into resorufin, a measurable fluorescent compound whose quantity correlates with the amount of living cells. None of the compounds tested were cytotoxic for HeLa cells; in fact, an increase in cell proliferation was observed for all three compounds (Fig. S5a†). However, optical images of the same cultures after compound removal demonstrated that the cell density was comparable to that of the control cultures (Fig. S5b†). This means that the compounds may increase the metabolic activity of the cells, but not their proliferation, or generate some kind of interference with the Alamar Blue technique, as has already been reported with both resazurin and MTT assays.27,28
Cell internalization of the compounds was assessed by confocal microscopy. Although the laser used was in the limit of the compound absorption range (300–400 nm), all compounds were successfully detected inside the cells, showing a fluorescence spotted pattern in the cytosol (Fig. 4a). To better appreciate the differences in the fluorescence intensity among the compounds, a greyscale (Fig. 4b) and a colour palette scale (Fig. 4c) were also used. In the greyscale, black and white colours indicate, respectively, the minimum and maximum fluorescence intensity, whereas the different grey colours represent intermediate intensities (Fig. 4b). In the colour palette scale (Fig. 4c), the intermediate intensities between the black and white ends are transformed into different colours (blue, red, orange and yellow) to easily visualize the differences in the fluorescence intensity. Notably, the fluorescence emission of the di-iodo derivative 6 was much greater than the mono-iodo and non-iodine derivatives 5 and 4, respectively, which suggests that the presence of two iodo groups leads to a more efficient transport across the plasma membrane and a better cellular uptake of the m-carborane anthracene dyads by HeLa cells.
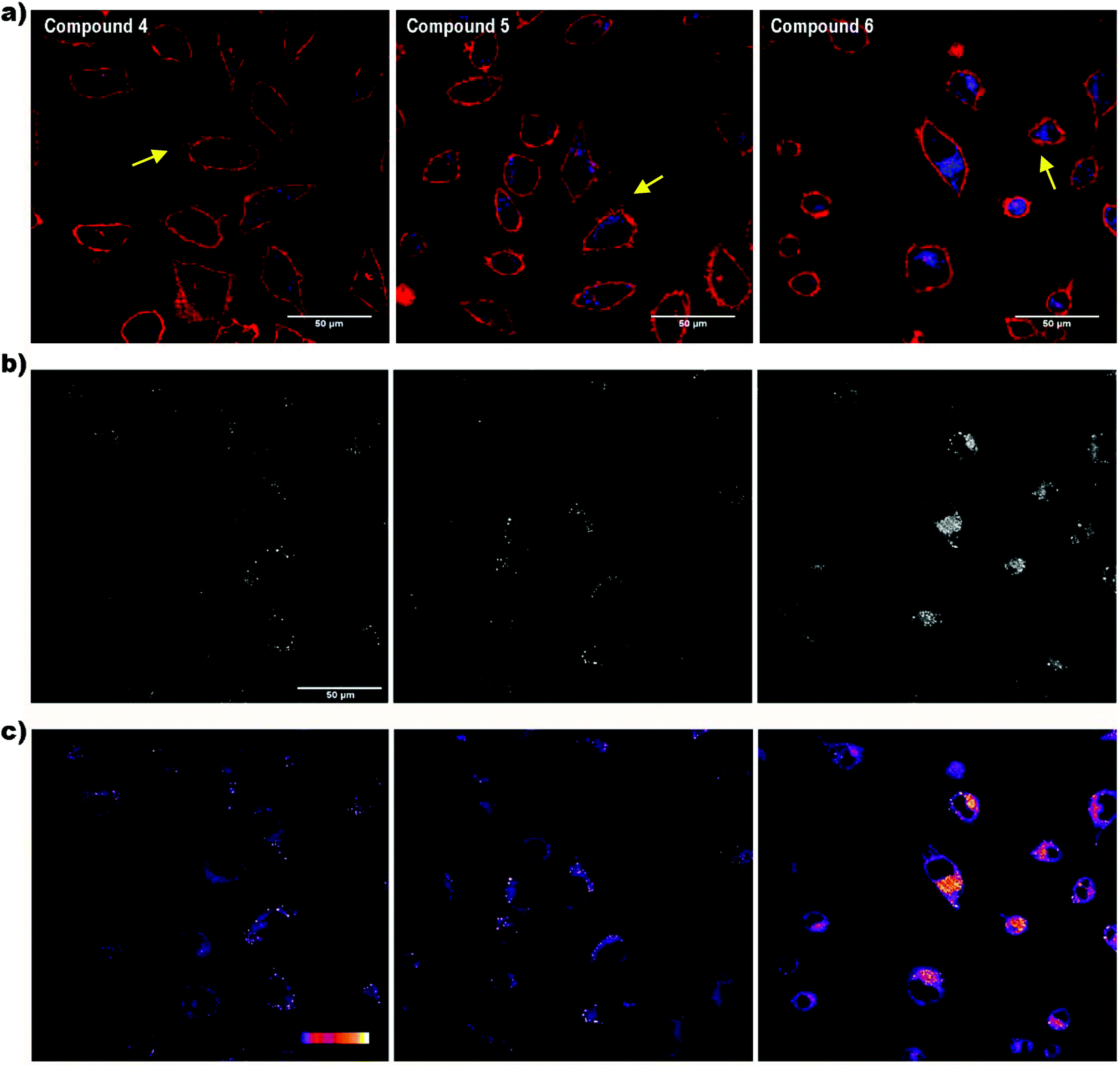 | ||
| Fig. 4 Cellular uptake of compounds 4, 5 and 6 by confocal microscopy. (a) Fluorescence intensity shown with a false colour assigned accordingly with the emission wavelength of the fluorochromes used. Plasma membrane (red); compounds (blue); blebs of the plasma membrane (yellow arrows). (b) Fluorescence intensity in a greyscale. (c) Fluorescence intensity transformed into a colour palette scale, in which blue and white colours represent the lowest and highest fluorescence intensity, respectively. Scale bar: 50 μm. | ||
The effect of iodine on the cell membrane transportability of fluorescent polymers has been recently reported, demonstrating that the introduction of iodo groups is able to enhance the cellular internalization of fluorescent probes, which could provide a novel strategy to improve the cellular uptake of organic molecules for drug delivery and cell tracking.29 It was suggested that the nature of halogen bonding plays a crucial role in plasma membrane transport in mammalian cells. Moreover, the influence of iodine on cell biological effects was previously discussed by our group for the case of the anionic boron cluster [3,3-Co(C2B9H11)2]− (COSAN) derivatives, where it was demonstrated that the cellular uptake for [3,3′-Co(8-I-C2B9H10)2]− (I2-COSAN) was higher than that for COSAN.30 In this case, the lipophilicity of I2-COSAN was approximately 3.5 times greater than that of COSAN, which suggests a higher affinity for the lipid environment.31 This higher lipophilicity is also consistent with the results obtained on artificial membranes, where the rate-limiting factor for COSAN and I2-COSAN translocation is due to partitioning between the lipid and water phase.30 The slower permeation rate seen for I2-COSAN could be due to its greater affinity for lipids.31 Altogether, the increase of lipophilicity with the iodo content and the improvement of cellular internalization would explain why compound 6 is better internalized by cells than compounds 4 and 5.
Regarding the laser used to excite the compounds, when living cells were illuminated for a few seconds with this laser, bubbles began to appear in the plasma membrane almost immediately (Fig. 4a, yellow arrows). The energy of the 405 nm laser is very potent and, probably, it causes cell damage in living cells visualized as plasma membrane blebs. Although these compounds would not be advisable for in vivo studies, they could be appropriate dyes for fixed cells.
On the other hand, the spotted fluorescence pattern of the compounds within the cell resembles that of the endosomes/lysosomes. Compound 6, the best internalized, was chosen to analyse whether the compound internalization was done through plasma membrane diffusion or by a mechanism of endocytosis, an energy-dependent (ATP consuming) process in which extracellular molecules are internalized through early-endosome vesicles which mature to late-endosomes and finally into lysosomes. For this purpose, the cells were incubated for 4 h with compound 6 either under standard conditions (37 °C) or at 4 °C, a temperature in which energy-dependent processes are arrested. Then, the fluorescence emission was assessed by confocal microscopy. At 37 °C a fluorescent spotted pattern typical of endosome/lysosome labelling was observed, indicating that 6 was, probably, inside some kinds of vesicles (Fig. 5a and b). At 4 °C the spotted pattern was also present, but the fluorescence intensity obtained was very low (Fig. 5c and d). The stability of compound 6 at 4 °C was also studied to rule out that the low intensity of the signal observed was due to a decrease in the fluorescence emission at this temperature (Fig. S6 and S7†).
 | ||
| Fig. 5 Cellular uptake of 6 by confocal microscopy at different temperatures. Internalization is considerable at 37 °C (a and b) but inefficient at 4 °C (c and d). Upper panel: Fluorescence intensity in a greyscale. Lower panel: Fluorescence intensity shown as a colour palette scale. Scale bar: 10 μm. | ||
The spotted pattern and the internalization arrest observed at 4 °C suggested that compound 6 enters the cell through endocytosis. In order to find this, new experiments were performed. It is well-known that Transferrin (Tf), an iron binding protein that facilitates iron uptake in cells, is internalized by endocytosis. Therefore, we analysed the cellular uptake of Transferrin-Alexa fluor 488 (Tf-A488) at 37 and 4 °C as a control of ATP-dependent transport inhibition at low temperature. Under standard conditions, this protein binds to its receptor located in the plasma membrane and is internalized by endocytosis showing the typical spotted pattern (Fig. 6a and b). However, when the same experiment was performed at 4 °C, the amount of Tf-A488 within the cells decreased considerably, being mainly located at the plasma membrane (Fig. 6c and d). This suggests that Tf-A488 can couple with its plasma membrane receptor, but the lack of ATP production prevents its internalization. To demonstrate that the low temperature did not promote cell death (which could also explain the low internalization), one of the cultures were kept at 4 °C for 3 h and then at 37 °C for 1 h, and in this case, the endocytosis was recovered (Fig. 6e and f). The obtained results showed that cellular uptake was considerably ineffective in all cultures incubated at 4 °C (compound 6 or Tf-A488), suggesting that endocytosis is the major pathway for the internalization of 6, but other mechanisms of transport could also be involved, since a low signal was detected within the cells at 4 °C.
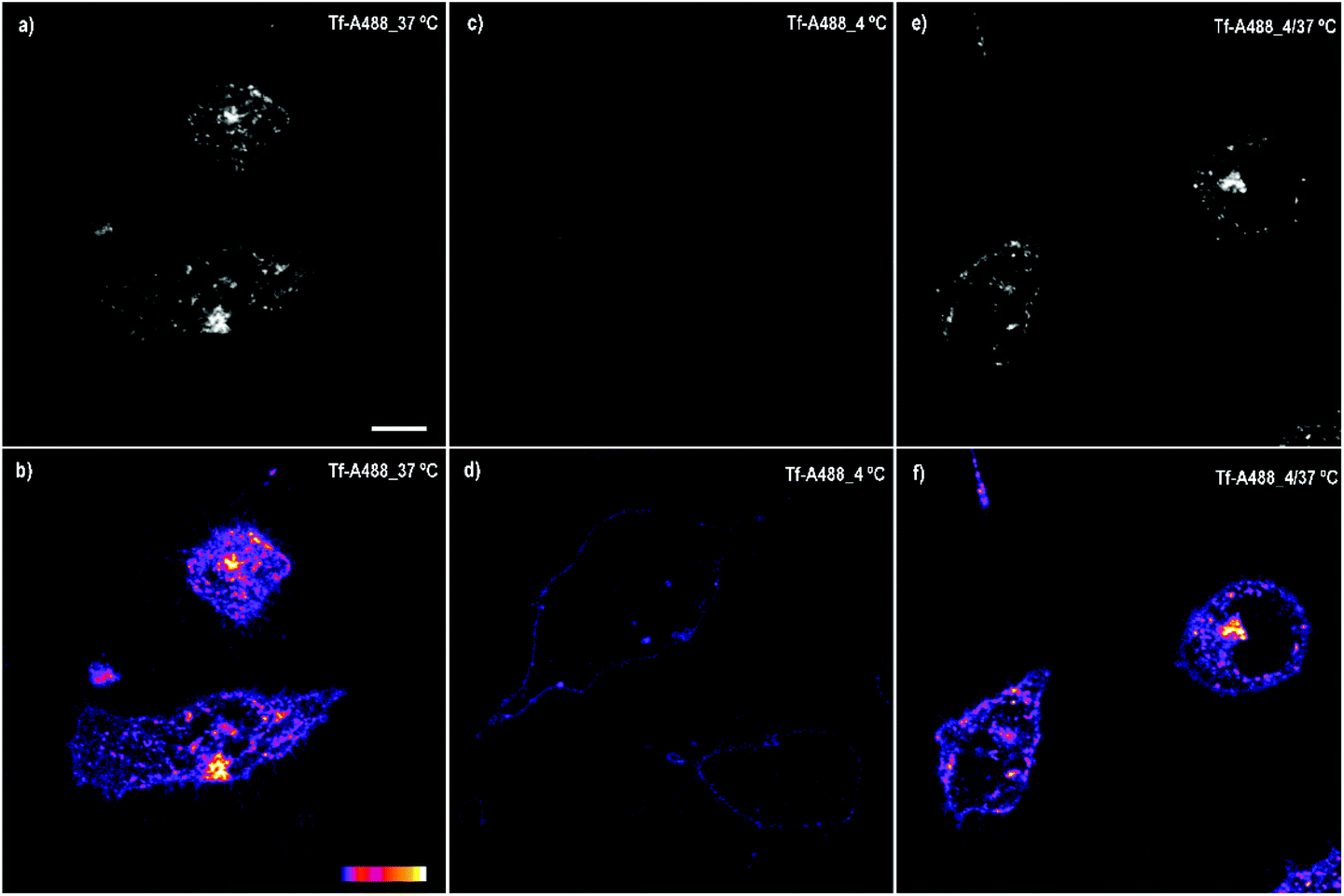 | ||
| Fig. 6 Cellular uptake of Tf-A488 by confocal microscopy at different temperatures. Internalization is considerable at 37 °C (a and b) but inefficient at 4 °C (c and d). Internalization can be recovered switching the temperature to standard conditions (e and f). Upper panel: Fluorescence intensity in a greyscale. Lower panel: Fluorescence intensity transformed into a colour palette scale. Scale bar: 10 μm. | ||
Finally, to confirm that compound 6 is internalized via endocytosis, colocalization experiments were performed, specifically using early-endosome and lysosome markers (Fig. 7). As expected, 6 is internalized via endocytosis. After 4 h of incubation 88.19% of the compound is colocalized with lysosomes, while only 22.16% with endosomes, which means that after 4 h most of the products had already reached the lysosomes. Again, false colours were assigned to fluorescence intensity to better visualize the organelles and the compound. In this case, green colour was assigned to compound 6 and red colour to organelles to obtain an orange colour when colocalization occurs.
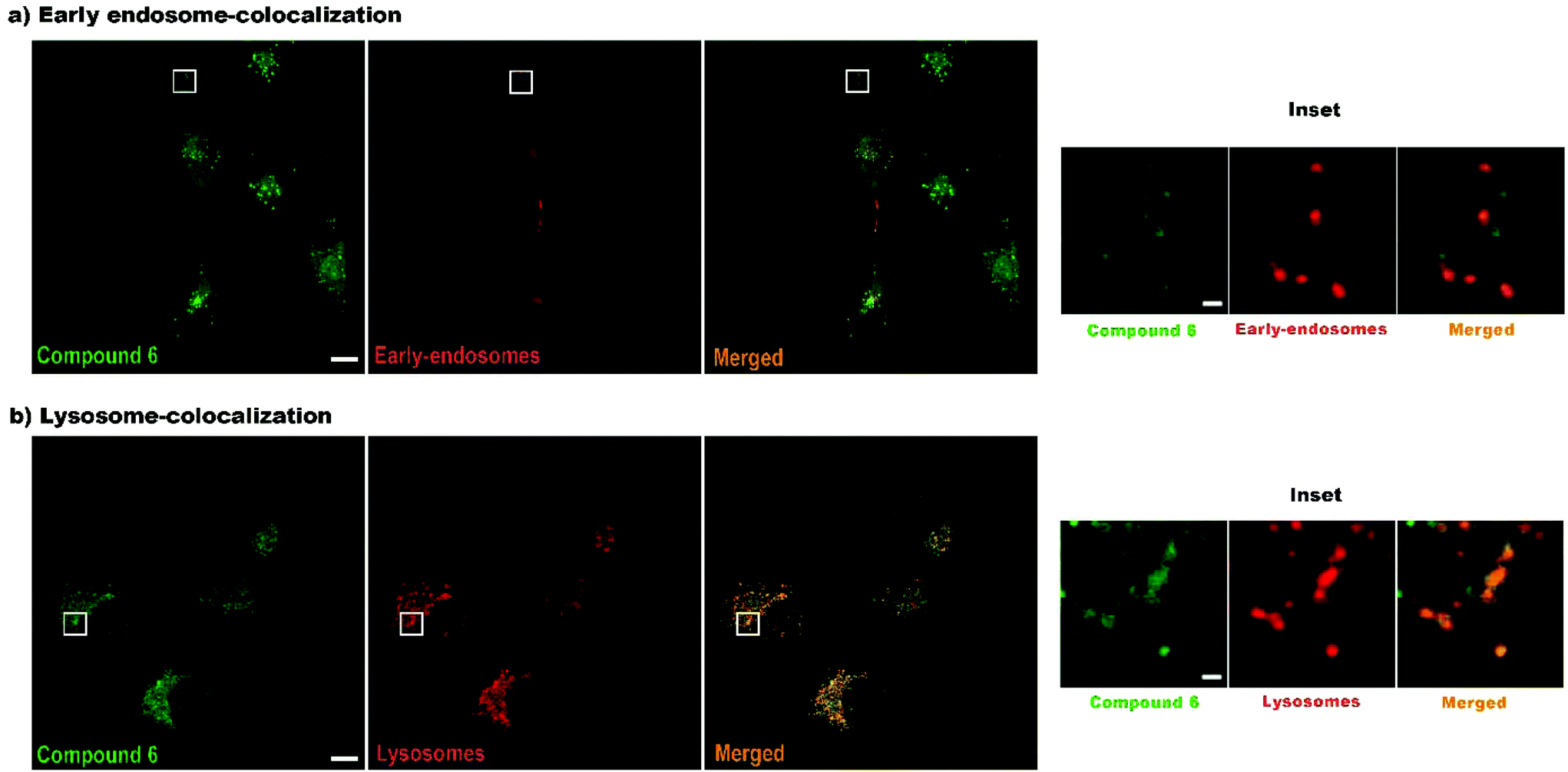 | ||
| Fig. 7 Colocalization of compound 6 with endosomes (a) and lysosomes (b). Compound 6 in green. Endosome/lysosome in red. Merged images showing colocalization in orange. Scale bar: 10 μm. Inset scale bar: 1 μm. | ||
Conclusions
A set of three m-carborane–anthracene dyads has been successfully synthesized by the monosubstitution of the m-carborane at one Ccluster; moreover, one or two hydrogens from the B–H groups have been substituted by iodine atoms. Remarkably, by linking m-carborane to the well-known anthracene dye produces an exceptional enhancement of the fluorescence properties in the final molecule, with fluorescence quantum yield values near 100% in solution for all of them. Noticeably, these values are much higher than those previously obtained for disubstituted m-carborane–anthracene triads, and clearly, the presence of the iodo units does not have any influence on the PL properties of compounds. These results indicate that our dyads are extremely good emitters in solution due to the free rotation of the anthracene linked to the Cc, as it is confirmed by TD-DFT calculations. Notably, these dyads are also good emitters in the aggregate state, which could be ascribed to the presence of CH2, particularly for 4, and the iodo units in 5 and 6, which prevent π–π stacking. Crystal packing, fingerprint plot analysis, and TD-DFT calculations for the three compounds support this hypothesis. Among the different compounds tested, compound 6 is the best-internalized compound by HeLa cells, the endocytic process being the main pathway observed to cross the plasma cell membrane. The high fluorescence intensity emitted, as well as the blue color emission, make this compound a potential agent to be applied in bioimaging studies with fixed cells.Experimental
Instrumentation
Elemental analyses were performed using a Carlo Erba EA1108 microanalyzer. ATR-IR spectra were recorded on a high-resolution JASCO FT/IR-4700 spectrometer. The 1H NMR (300.13 MHz), 11B{1H} (96.29 MHz) and 13C{1H} NMR (75.47 MHz) spectra were recorded on a Bruker ARX 300 spectrometer. All NMR spectra were recorded in CDCl3 solutions at 25 °C. Chemical shift values for 11B{1H} NMR spectra were referenced to external BF3·OEt2, and those for 1H and 13C{1H} NMR were referenced to SiMe4 (TMS). Chemical shifts are reported in units of parts per million downfield from the reference, and all coupling constants are reported in hertz. UV-Vis spectra were recorded using a VARIANT Cary 5 UV-Vis-NIR spectrophotometer, using spectroscopic grade THF (Sigma-Aldrich), in a normal quartz cuvette having 1 cm path length, for different solutions for each compound in the range of 10−4 to 10−5 M in order to calculate the molar extinction coefficients (ε). The fluorescence emission spectra and excitation spectra for all samples were recorded using a Varian Cary Eclipse fluorescence spectrometer. No fluorescent contaminants were detected on excitation in the wavelength region of experimental interest. The fluorescence quantum yields were determined by a “single point method” and repeated three times with a similar optical density for reproducibility,32 against quinine sulfate in 0.5 M aqueous sulfuric acid with ϕF = 0.54 as a standard.33 For the suspensions in THF/water (1/99, v/v) the refractive index was assumed to be that of pure water (1.33). Confocal laser scanning microscopy (CLSM, Olympus) was used to analyze the cell internalization. The images obtained were processed with ImageJ software.Materials
All reactions were performed under an atmosphere of dinitrogen employing standard Schlenk techniques. Tetrahydrofuran was purchased from Merck and distilled from sodium benzophenone prior to use. Commercial grade diethyl ether, hexane, petroleum ether, n-heptane, chloroform and dichloromethane were used without further purification. 1,7-closo-C2B10H12 (1) was supplied from KatChem Ltd (Prague) and used as received. Compounds 9-I-1,7-dicarba-closo-dodecaborane (2) and 9,10-I2-1,7-dicarba-closo-dodecaborane (3) were synthesized according to the literature.21n-BuLi solution (1.6 M in hexane) was purchased from Aldrich and 9-(chloromethyl)anthracene was purchased from Alfa Aesar. Cell Mask Orange was purchased from Thermo Fisher.X-ray single-crystal structure determination
Measured crystals were prepared under inert conditions immersed in perfluoropolyether as protecting oil for manipulation. Suitable crystals were mounted on MiTeGen Micromounts™ and these samples were used for data collection. Data were collected with a Bruker D8 Venture diffractometer. The data were processed with the APEX3 suite [Bruker, Bruker AXS Inc. V2016.1, 2016, Madison, Wisconsin, USA]. The structures were solved by direct methods,34 which revealed the position of all non-hydrogen atoms. These atoms were refined on F2 by a full-matrix least-squares procedure using anisotropic displacement parameters. All hydrogen atoms were located in difference Fourier maps and included as fixed contributions riding on attached atoms with isotropic thermal displacement parameters 1.2 times those of the respective atom. Crystallographic data for the structure of compounds 4, 5 and 6 have been deposited with the Cambridge Crystallographic Data Center as supplementary publication no. CCDC 1907569–1907571.† Geometric calculations and molecular graphics were performed with Mercury.35 Additional crystal data are shown in Table 1.Biological assays
Compounds 4–6 were dissolved and filtered following the same protocol as that in a previous work,26 obtaining a final compound concentration of 10 μM and 0.5% of DMSO (Sigma/Aldrich).26 HeLa cells, which were routinely cultured under standard conditions, were used to analyse the cytotoxicity, the cellular uptake and the internalization pathway.26,27Cytotoxicity
The Alamar Blue assay (Invitrogen) was used to assess the cytotoxicity of the compounds. To perform these experiments, 6 × 104 cells per ml were seeded into 24 well plates and the day after they were incubated with each compound at 10 μM for 24 h. Then, the compounds were removed and the cells were rinsed with 500 μl of HBSS and incubated with a fresh medium containing 10% of Alamar Blue for 3 h at 37 °C protected from light. Finally, the medium with Alamar Blue was collected and the fluorescence intensity was measured with a Cary Eclipse Fluorescence Spectrophotometer (λex = 560; λem = 590; Agilent). Experiments were performed in triplicate and the results were normalized with control cultures (non-treated cells cultured under the same conditions without any compounds and considered as 100% of cell viability).Cellular uptake
HeLa cells (2 × 105 cells) were seeded into glass bottom culture dishes (MatTek) and after 24 h, they were exposed to 1 ml of each compound at 10 μM for 4 h at 37 °C. The cell cultures were rinsed twice with sterile phosphate buffered solution (PBS from Biowest), and the plasma membrane was stained with 0.5 μl ml−1 of Cell Mask Orange (Thermo Fisher). Finally, the cells were observed under Confocal Laser Scanning Microscopy (CLSM) (Olympus Fluoview 1000) and the images were processed by ImageJ/Fiji software. Fluorescence settings: product 6 (λex = 405 nm; λem = 425–475 nm) and plasma membrane (λex = 559 nm; λem = 570–625 nm).Internalization analysis
For each experiment, HeLa cells were seeded into glass bottom culture dishes (MatTek) at a density of 2 × 105 cells and maintained under standard culture conditions for 24 h. The cells were incubated with 1 ml of compound 6 at 10 μM for 4 h at two different temperatures: 37 °C and 4 °C. Additionally, one culture was incubated for 4 h at 37 °C and after removing the compound, it was maintained for an additional 1 h at 4 °C to assess the effect of temperature on the fluorescence intensity.As a control of endocytosis, the cells were incubated with 20 μg ml−1 of Transferrin-Alexa 488 (Tf-A488 from Life Technologies) for 4 h at the same temperatures (37 °C and 4 °C). In addition, one culture with Tf-A488 was incubated for 3 h at 4 °C and then for 1 h at 37 °C to confirm that endocytosis restarts when optimal conditions are recovered. In experiments carried out at low temperature, the cells were kept at 4 °C during 1 h prior to the addition of 6 or Tf-A488 to arrest their enzymatic activity. Finally, after incubation, the compounds were rinsed with PBS and observed under a CLSM (Olympus Fluoview 1000). Non-treated cells were also analysed by confocal microscopy using the same settings as a control for autofluorescence (Fig. S6†).
Colocalization
Cells (2 × 105) were seeded into glass bottom culture dishes. Two different cell cultures were prepared to label endosomes and lysosomes separately. To label endosomes, upon cell attachment, 10 μl of CellLight™ Early Endosomes-RFP, BacMam 2.0 (Invitrogen), was added to the cell culture and the cells were maintained overnight under standard culture conditions. Lysosomes were labelled with LysoTracker Red (1:500, Invitrogen) during 30 min and rinsed prior to observation. Compound 6, at a concentration of 5 μM, was added to the culture and placed at 37 °C. After 4 h of incubation, product 6 was rinsed and replaced with fresh medium. The images were acquired with a Leica SP5 CLSM. Endosome and lysosome colocalization with the product was quantified by Imaris software. Fluorescence settings: product 6 (λex = 405 nm; λem = 423–516 nm); early endosomes (λex = 561 nm; λem = 581–717 nm,); and lysosomes (λex = 561 nm; λem = 581–717 nm).
Synthesis of 4
A dry 250 mL round-bottomed flask equipped with a magnetic stirring bar was charged under nitrogen with a solution of 1,7-C2B10H12 (1) (0.600 g, 4.17 mmol) in THF (80 mL) at 0 °C. Then, a solution of 1.6 M n-BuLi in hexanes (2.60 mL, 4.17 mmol) was added dropwise to the mixture, which was allowed to stir for 1 h at room temperature and cooled again at 0 °C. A solution of 9-chloromethyl anthracene (0.964 g, 4.17 mmol) in THF (30 mL) was then added dropwise to the mixture under vigorous stirring. Then it was stirred for a half an hour at room temperature and heated to reflux overnight. After that, the solvent was removed under vacuum and the residue was quenched with H2O (15 mL), transferred to a separating funnel, and extracted with Et2O (3 × 10 mL). The organic layer was dried over MgSO4 and the volatiles were reduced under vacuum. The orange oil residue was purified by preparative layer chromatography (dichloromethane/hexane 10:90) to give 4 as a yellowish white solid. Yield: 0.722 g, 52%. Crystals suitable for X-ray analysis were obtained by slow evaporation of a solution of 4 in chloroform/n-heptane. 1H NMR, δ (ppm): 8.44 (s, 1H, C14H9), 8.19 (d, 3J(H,H) = 9 Hz, 2H, C14H9), 8.02 (d, 3J(H,H) = 9 Hz, 2H, C14H9), 7.61–7.55 (m, 2H, C14H9), 7.49 (t, 3J(H,H) = 7.5 Hz, 2H, C14H9), 4.29 (s, 2H, CH2), 2.75 (s, 1H, CcH); 11B{1H} NMR, δ (ppm): −3.66 (s, 2B), −10.80 (s, 4B), −13.99 (s, 2B), −15.58 (s, 2B); 13C{1H} NMR, δ (ppm): 131.53 (s, C14H9), 130.94 (s, C14H9), 129.38 (s, C14H9), 128.33 (s, C14H9), 126.35 (s, C14H9), 125.13 (s, C14H9), 124.85 (s, C14H9), 55.10 (s, Cc–H), 33.88 (s, CH2); ATR-IR (cm−1): ν = 3049 (Car–H), 2591, 2577 (B–H), 1625 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C); elemental analysis calcd (%) for C17H22B10: C, 61.05; H, 6.63. Found: C, 60.98; H, 6.77.
C); elemental analysis calcd (%) for C17H22B10: C, 61.05; H, 6.63. Found: C, 60.98; H, 6.77.
Synthesis of 5
The procedure was the same as that for 4, but using a solution of 9-I-1,7-C2B10H12 (2) (0.819 g, 3.032 mmol) in THF (70 mL), 1.6 M n-BuLi in hexanes (1.90 mL, 3.04 mmol) and a solution of 9-chloromethyl anthracene (0.702 g, 3.034 mmol) in THF (25 mL). After extraction with 3 × 10 mL of brine/CH2Cl2 the orange oil was purified by preparative layer chromatography (dichloromethane/petroleum ether 20:80) to give 5 as a yellowish solid. Yield: 0.754 g, 54%. Crystals suitable for X-ray analysis were obtained by slow evaporation of a solution of 5 in chloroform. 1H NMR, δ (ppm): 8.44 (s, 1H, C14H9), 8.13 (dd, 3J(H,H) = 9 Hz, 3J(H,H) = 6 Hz, 2H, C14H9), 8.02 (d, 3J(H,H) = 9 Hz, 2H, C14H9), 7.61–7.56 (m, 2H, C14H9), 7.50 (t, 3J(H,H) = 7.5 Hz, 2H, C14H9), 4.29 (s, 2H, CH2), 2.84 (s, 1H, CcH); 11B{1H} NMR, δ (ppm): −2.65 (s, 1B), −8.93 (s, 4B), −12.82 (s, 1B), −13.68 (s, 1B), −15.39 (s, 1B), −17.61 (s, 1B), −23.87 (s, 1B); 13C{1H} NMR, δ (ppm): 131.47 (s, C14H9), 130.80 (s, C14H9), 129.47 (s, C14H9), 128.71 (s, C14H9), 128.62 (s, C14H9), 126.62 (s, C14H9), 125.20 (s, C14H9), 124.45 (s, C14H9), 56.07 (s, Cc–H), 33.72 (s, CH2); ATR-IR (cm−1): ν = 3048, 2929, 2852 (Car–H), 2595, 2572, 2556 (B–H), 1626 (CC); elemental analysis calcd (%) for C17H21B10I: C, 44.35; H, 4.60. Found: C, 44.42; H, 4.61.
Synthesis of 6
The procedure was the same as that for 4, but using a solution of 9,10-I2-1,7-dicarba-closo-dodecaborane (3) (0.760 g, 1.919 mmol) in THF (80 mL), 1.6 M n-BuLi in hexanes (1.20 mL, 1.920 mmol) and a solution of 9-chloromethyl anthracene (0.444 g, 1.919 mmol) in THF (20 mL). After extraction with 3 × 10 mL of brine/CH2Cl2 the orange oil was purified by preparative layer chromatography (dichloromethane/petroleum ether 20:80) to give 6 as a yellow solid. Yield: 0.461 g, 41%. Crystals suitable for X-ray analysis were obtained by slow evaporation of a solution of 6 in dichloromethane. 1H NMR, δ (ppm): 8.47 (s, 1H, C14H9), 8.09 (d, 3J(H,H) = 9 Hz, 2H, C14H9), 8.03 (d, 3J(H,H) = 9 Hz, 2H, C14H9), 7.60 (t, 3J(H,H) = 7.5 Hz, 2H, C14H9), 7.50 (t, 3J(H,H) = 7.5 Hz, 2H, C14H9), 4.31 (s, 2H, CH2), 2.92 (s, 1H, CcH); 11B{1H} NMR, δ (ppm): −1.62 (s, 1B), −7.56 (s, 1B), −9.08 (s, 2B), −12.77 (s, 2B), −17.47 (s, 2B), −21.15 (s, 2B); 13C{1H} NMR, δ (ppm): 131.49 (s, C14H9), 130.76 (s, C14H9), 129.60 (s, C14H9), 128.93 (s, C14H9), 128.14 (s, C14H9), 126.91 (s, C14H9), 125.31 (s, C14H9), 124.17 (s, C14H9), 56.68 (s, Cc–H), 33.69 (s, CH2); ATR-IR (cm−1): ν = 3035, 2962, 2929, 2856 (Car–H), 2609 (B–H), 1623 (CC); elemental analysis calcd (%) for C17H20B10I2: C, 34.83; H, 3.44. Found: C, 34.68; H, 3.43.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The work was supported by the Spanish Ministerio de Economía y Competitividad, MINECO (CTQ2016-75150-R, MAT2017-86357-C3-3-R and “Severo Ochoa” Program for Centers of Excellence in R&D SEV-2015-0496) and Generalitat de Catalunya (2017/SGR/1720, 2017/SGR/503). Z. K. is grateful for the general support of the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement MSCA-IF-2016-751587. D. Ch.-L. acknowledges funding from the Intramural CSIC (201730I019) project. Theoretical calculations have been achieved using computers from the Supercomputing Centre of Catalonia (CESCA). The authors would like to thank the staff at the Servei de Microscòpia de la Universitat Autònoma de Barcelona.References
- M. Yoshizawa and J. K. Klosterman, Chem. Soc. Rev., 2014, 43, 1885–1898 RSC.
- (a) J. Liu, L. Jiang and W. P. Hu, Prog. Chem., 2009, 21, 2568–2577 CAS; (b) M. Y. Chen, Y. Zhao, L. J. Yan, S. Yang, Y. N. Zhu, I. Murtaza, G. F. He, H. Meng and W. Huang, Angew. Chem., Int. Ed., 2017, 56, 722–727 CrossRef CAS PubMed; (c) Y. Takaki, Y. Wakayama, Y. Ishiguro, R. Hayakawa, M. Yamagishi, T. Okamoto, J. Takeya, K. Yoza and K. Kobayashi, Chem. Lett., 2016, 45, 1403–1405 CrossRef CAS; (d) M. Y. Chen, Y. Zhao, L. J. Yan, S. Yang, Y. N. Zhu, I. M. Chen, L. Yan, Y. Zhao, I. Murtaza, H. Meng and W. Huang, J. Mater. Chem. C, 2018, 6, 7416–7444 RSC.
- S. Erdemir and O. Kocyigit, Talanta, 2016, 158, 63–69 CrossRef CAS PubMed.
- (a) J. Chen, A. Neels and K. M. Fromm, Chem. Commun., 2010, 46, 8282–8284 RSC; (b) R. Iwaura, H. Yui, Y. Someya and M. Ohnishi-Kameyama, J. Photochem. Photobiol., B, 2014, 130, 199–204 CrossRef CAS PubMed; (c) C. Zhaoa, X. Caia, Z. Maa, J. Shia, L. Xub and H. Wanga, J. Photochem. Photobiol., A, 2018, 355, 318–325 CrossRef; (d) Y. Gao, H. Liu, S. Zhang, Q. Gu, Y. Shen, Y. Gea and B. Yang, Phys. Chem. Chem. Phys., 2018, 20, 12129–12137 RSC.
- (a) T. Hinoue, Y. Shigenoi, M. Sugino, Y. Mizobe, I. Hisaki, M. Miyata and N. Tohnai, Chem. – Eur. J., 2012, 18, 4634–4643 CrossRef CAS PubMed; (b) S. Yamane, Y. Sagara and T. Kato, Chem. Commun., 2013, 49, 3839–3841 RSC.
- (a) V. I. Bregadze, Chem. Rev., 1992, 92, 209–223 CrossRef CAS; (b) F. Teixidor, C. Viñas, A. Demonceau and R. Núnez, Pure Appl. Chem., 2003, 75, 1305–1313 CAS; (c) C. Viñas and F. Teixidor, Science of Synthesis, Thieme Stuttgart, 2005, vol. 6, pp. 1235–1275 Search PubMed; (d) M. Scholz and E. Hey Hawkins, Chem. Rev., 2011, 111, 7035–7062 CrossRef CAS PubMed; (e) F. Issa, M. Kassiou and L. M. Rendina, Chem. Rev., 2011, 111, 5701–5722 CrossRef CAS PubMed; (f) N. S. Hosmane, Boron Science: New Technologies and Applications, Taylor & Francis, 2011 Search PubMed; (g) Z. J. Yao and G. X. Jin, Coord. Chem. Rev., 2013, 257, 2522–2535 CrossRef CAS; (h) A. M. Spokoyny, Pure Appl. Chem., 2013, 85, 903–919 CAS; (i) J. Zhang and Z. Xie, Acc. Chem. Res., 2014, 47, 1623–1633 CrossRef CAS PubMed; (j) R. N. Grimes, Carboranes, Academic Press, US, 3rd edn, 2016 Search PubMed; (k) N. S. Hosmane and R. Eagling, in Handbook of Boron Science with Applications in Organometallics, Catalysis, Materials and Medicine, World Scientific, Singapore, 2019 Search PubMed.
- (a) J. Poater, M. Solà, C. Viñas and F. Teixidor, Angew. Chem., Int. Ed., 2014, 53, 12191–12195 CrossRef CAS PubMed; (b) J. Poater, M. Solà, C. Viñas and F. Teixidor, Chem. – Eur. J., 2013, 19, 4372–4372 CrossRef.
- (a) J. Cabrera-González, A. Ferrer-Ugalde, S. Bhattacharyya, M. Chaari, F. Teixidor, J. Gierschner and R. Núñez, J. Mater. Chem. C, 2017, 5, 10211–10219 RSC; (b) A. González-Campo, A. Ferrer-Ugalde, C. Viñas, F. Teixidor, R. Sillanpää, J. Rodríguez-Romero, R. Santillan, N. Farfán and R. Núñez, Chem. – Eur. J., 2013, 19, 6299–6312 CrossRef PubMed; (c) A. Ferrer-Ugalde, E. J. Juárez Pérez, F. Teixidor, C. Viñas and R. Núñez, Chem. – Eur. J., 2013, 19, 17021–17030 CrossRef CAS PubMed.
- (a) J. Plešek, Chem. Rev., 1992, 92, 269–278 CrossRef; (b) I. B. Sivaev and V. V. Bregadze, Eur. J. Inorg. Chem., 2009, 1433–1450 CrossRef CAS; (c) C. Viñas, Future Med. Chem., 2013, 5, 617–619 CrossRef PubMed; (d) M. A. Soriano-Ursúa, B. C. Das and J. G. Trujillo-Ferrara, Expert Opin. Ther. Pat., 2014, 24, 485–500 CrossRef PubMed; (e) Z. J. Leśnikowski, J. Med. Chem., 2016, 59, 7738–7758 CrossRef PubMed; (f) G. Calabrese, A. Daou, A. Rova, E. Tseligka, I. S. Vizirianakis, D. G. Fatouros and J. Tsibouklis, Med. Chem. Commun., 2017, 8, 67–72 RSC.
- R. Núñez, I. Romero, F. Teixidor and C. Viñas, Chem. Soc. Rev., 2016, 45, 5137–5434 RSC.
- (a) R. Núñez, P. Farràs, F. Teixidor, C. Viñas, R. Sillanpää and R. Kivekäs, Angew. Chem., Int. Ed., 2006, 45, 1270–1272 CrossRef PubMed; (b) A. M. Spokoyny, C. W. Machan, D. J. Clingerman, M. S. Rosen, M. J. Wiester, R. D. Kennedy, C. L. Stern, A. A. Sarjeant and C. A. Mirkin, Nat. Chem., 2011, 3, 590–596 CrossRef CAS PubMed.
- J. Llop, C. Viñas, F. Teixidor, L. Victori, R. Kivekäs and R. Sillanpää, Organometallics, 2001, 20, 4024–4030 CrossRef CAS.
- (a) R. Núñez, M. Tarrés, A. Ferrer-Ugalde, F. F. de Biani and F. Teixidor, Chem. Rev., 2016, 116, 14307–14378 CrossRef PubMed; (b) S. Mukherjee and P. Thilagar, Chem. Commun., 2016, 52, 1070–1093 RSC.
- (a) K. R. Wee, Y. J. Cho, S. Jeong, S. Kwon, J. D. Lee, I. H. Suh and S. O. Kang, J. Am. Chem. Soc., 2012, 134, 17982–17990 CrossRef CAS PubMed; (b) L. Zhu, W. Lv, S. Liu, H. Yan, Q. Zhao and W. Huang, Chem. Commun., 2013, 49, 10638–10640 RSC; (c) J. Kahlert, L. Bohling, A. Brockhinke, H. G. Stammler, B. Neumann, L. M. Rendina, P. J. Low, L. Weber and M. A. Fox, Dalton Trans., 2015, 44, 9766–9781 RSC; (d) D. Tu, P. Leong, Z. Li, R. Hu, C. Shi, K. Y. Zhang, H. Yan and Q. Zhao, Chem. Commun., 2016, 52, 12494–12497 RSC; (e) Y. Kim, S. Park, Y. H. Lee, J. Jung, S. Yoo and M. H. Lee, Inorg. Chem., 2016, 55, 909–917 CrossRef CAS PubMed; (f) R. Furue, T. Nishimoto, I. S. Park, J. Lee and T. Yasuda, Angew. Chem., Int. Ed., 2016, 55, 7171–7175 CrossRef CAS PubMed; (g) D. S. Tuo, P. Leong, S. Guo, H. Yan, C. S. Lu and Q. Zhao, Angew. Chem., Int. Ed., 2017, 56, 11370–11374 CrossRef PubMed; (h) H. Naito, K. Nishino, Y. Morisaki, K. Tanaka and Y. Chujo, J. Mater. Chem. C, 2017, 5, 10047–10054 RSC.
- (a) H. Naito, Y. Morisaki and Y. Chujo, Angew. Chem., 2015, 127, 5173–5176 CrossRef; (b) Y. J. Cho, S. Y. Kim, M. Cho, W. S. Han, H. J. Son, D. W. Cho and S. O. Kang, Phys. Chem. Chem. Phys., 2016, 18, 9702–9708 RSC; (c) Z. Wang, P. Jiang, T. Wang, G. J. Moxey, M. P. Cifuentes, C. Zhang and M. G. Humphrey, Phys. Chem. Chem. Phys., 2016, 18, 15719–15726 RSC; (d) K. Tanaka, K. Nishino, S. Ito, H. Yamane, K. Suenaga, K. Hashimoto and Y. Chujo, Faraday Discuss., 2017, 196, 31–42 RSC; (e) H. Naito, K. Nishino, Y. Morisaki, K. Tanaka and Y. Chujo, Chem. – Asian J., 2017, 12, 2134–2138 CrossRef CAS PubMed; (f) K. Nishino, K. Uemura, M. Gon, K. Tanaka and Y. Chujo, Molecules, 2017, 22, 2009 CrossRef PubMed.
- (a) A. Ferrer-Ugalde, E. J. Juárez-Pérez, F. Teixidor, C. Viñas, R. Sillanpää, E. Pérez-Inestrosa and R. Núñez, Chem. – Eur. J., 2012, 18, 544–553 CrossRef CAS PubMed; (b) A. Ferrer-Ugalde, A. González-Campo, C. Viñas, J. Rodríguez-Romero, R. Santillan, N. Farfán, R. Sillanpää, A. Sousa-Pedrares, R. Núñez and F. Teixidor, Chem. – Eur. J., 2014, 20, 9940–9951 CrossRef CAS PubMed; (c) A. Ferrer-Ugalde, J. Cabrera-González, E. J. Juárez-Pérez, F. Teixidor, E. Pérez-Inestrosa, J. M. Montenegro, R. Sillanpää, M. Haukka and R. Núñez, Dalton Trans., 2017, 46, 2091–2104 RSC; (d) J. Cabrera-González, S. Bhattacharyya, B. Milián-Medina, F. Teixidor, N. Farfán, R. Arcos-Ramos, V. Vargas-Reyes, J. Gierschner and R. Núñez, Eur. J. Inorg. Chem., 2017, 4575–4580 CrossRef.
- J. Cabrera-González, C. Viñas, M. Haukka, S. Bhattacharyya, J. Gierschner and R. Núñez, Chem. – Eur. J., 2016, 22, 13588–13598 CrossRef PubMed.
- (a) H. Naito, Y. Morisaki and Y. Chujo, Angew. Chem., Int. Ed., 2015, 54, 5084–5087 CrossRef CAS PubMed; (b) H. Naito, K. Nishino, Y. Morisaki, K. Tanaka and Y. Chujo, Angew. Chem., Int. Ed., 2017, 56, 254–259 CrossRef CAS PubMed; (c) X. Wu, J. Guo, Y. Quan, W. Jia, D. Jia, Y. Chen and Z. Xie, J. Mater. Chem. C, 2018, 6, 4140–4149 RSC; (d) X. Wu, J. Guo, Y. Cao, J. Zhao, W. Jia, Y. Chen and D. Jia, Chem. Sci., 2018, 9, 5270–5277 RSC; (e) X. Wu, J. Guo, J. Zhao, W. Jia, D. Jia and H. Shan, Dyes Pigm., 2019, 162, 855–862 CrossRef CAS.
- M. Chaari, Z. Kelemen, J. Giner-Planas, F. Teixidor, D. Choquesillo-Lazarte, A. Ben Salah, C. Viñas and R. Núñez, J. Mater. Chem. C, 2018, 6, 11336–11347 RSC.
- A. R. Popescu, A. D. Musteti, A. Ferrer-Ugalde, C. Viñas, R. Núñez and F. Teixidor, Chem. – Eur. J., 2012, 18, 3174–3184 CrossRef CAS PubMed.
- M. A. Fox, PhD Thesis, University of Durham, 1991.
- (a) H. Y. V. Ching, S. Clifford, M. Bhadbhade, R. J. Clarke and L. M. Rendina, Chem. – Eur. J., 2012, 18, 14413–14425 CrossRef CAS PubMed; (b) H. Y. V. Ching, R. J. Clarke and L. M. Rendina, Inorg. Chem., 2013, 52, 10356–10367 CrossRef CAS PubMed; (c) M. Y. Tsang, F. Di Salvo, F. Teixidor, C. Viñas, J. Giner Planas, D. Choquesillo-Lazarte and N. Vanthuyne, Cryst. Growth Des., 2015, 15, 935–945 CrossRef CAS; (d) E. Oleshkevich, F. Teixidor, D. Choquesillo-Lazarte, R. Sillanpää and C. Viñas, Chem. – Eur. J., 2016, 22, 3665–3670 CrossRef CAS PubMed; (e) E. Oleshkevich, C. Viñas, I. Romero, D. Choquesillo-Lazarte, M. Haukka and F. Teixidor, Inorg. Chem., 2017, 56, 5502–5505 CrossRef CAS PubMed.
- A. V. Puga, F. Teixidor, R. Sillanpää, R. Kivekääs, M. Arca, G. Barberà and C. Viñas, Chem. – Eur. J., 2009, 15, 9755–9763 CrossRef CAS PubMed.
- M. Chaari, J. Cabrera-González, Z. Kelemen, C. Viñas, A. Ferrer-Ugalde, D. Choquesillo-Lazarte, A. Ben Salah, F. Teixidor and R. Núñez, J. Organomet. Chem., 2018, 865, 206–213 CrossRef CAS.
- R. Akatsuka, A. Momotake, Y. Shinohara, Y. Kanna, T. Sato, M. Moriyama, K. Takahashi, Y. Nishimura and T. Arai, J. Photochem. Photobiol., A, 2018, 355, 318–325 CrossRef.
- C. Bellomo, M. Chaari, J. Cabrera-González, M. Blangetti, C. Lombardi, A. Deagostino, C. Viñas, N. Gaztelumendi, C. Nogués, R. Nuñez and C. Prandi, Chem. – Eur. J., 2018, 24, 15622–15630 CrossRef CAS PubMed.
- M. Chaari, N. Gaztelumendi, J. Cabrera-González, P. Peixoto-Moledo, C. Viñas, E. Xochitiotzi-Flores, N. Farfán, A. Ben Salah, C. Nogués and R. Núñez, Bioconjugate Chem., 2018, 29, 1763–1773 CrossRef CAS PubMed.
- B. H. Neufeld, J. B. Tapia, A. Lutzke and M. M. Reynolds, Anal. Chem., 2018, 90, 6867–6876 CrossRef CAS PubMed.
- T. Wang, P. Yin, Y. Yang, W. Yin, S. Zhang, M. Yang, Y. Qin, Y. Ma, Z. Lei and H. Ma, ACS Sustainable Chem. Eng., 2019, 7, 6295–6303 CrossRef CAS.
- C. Verdiá-Báguena, A. Alcaraz, V. M. Aguilella, A. M. Cioran, S. Tachikawa, H. Nakamura, F. Teixidor and C. Viñas, Chem. Commun., 2014, 50, 6700 RSC.
- M. Tarrés, E. Canetta, E. Paul, J. Forbes, K. Azzouni, C. Viñas, F. Teixidor and A. J. Harwood, Sci. Rep., 2015, 5, 7804 CrossRef PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer US, Baltimore, Maryland, USA, 2007 Search PubMed.
- A. M. Brouwer, Pure Appl. Chem., 2011, 83, 2213–2228 CAS.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr, 2008, 64, 112–122 CrossRef CAS PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 11B{1H} NMR spectra, computational details, crystallographic data and Hirshfeld surface analyses for all the compounds. CCDC 1907569–1907571. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9bm00903e |
| This journal is © The Royal Society of Chemistry 2019 |