
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dual bioresponsive antibiotic and quorum sensing inhibitor combination nanoparticles for treatment of Pseudomonas aeruginosa biofilms in vitro and ex vivo†
Nishant
Singh‡
a,
Manuel
Romero‡
b,
Alessandra
Travanut
a,
Patricia F.
Monteiro
 a,
Elena
Jordana-Lluch§
c,
Kim R.
Hardie
c,
Paul
Williams
c,
Morgan R.
Alexander
a and
Cameron
Alexander
*a
a,
Elena
Jordana-Lluch§
c,
Kim R.
Hardie
c,
Paul
Williams
c,
Morgan R.
Alexander
a and
Cameron
Alexander
*a
aSchool of Pharmacy, University of Nottingham, University Park, Nottingham NG7 2RD, UK. E-mail: cameron.alexander@nottingham.ac.uk
bNational Biofilms Innovation Centre (NBIC), School of Life Sciences, University of Nottingham, Centre for Biomolecular Sciences, University Park, NG7 2RD, UK
cSchool of Life Sciences, University of Nottingham, Centre for Biomolecular Sciences, University Park, NG7 2RD, UK
First published on 29th July 2019
Abstract
Many debilitating infections result from persistent microbial biofilms that do not respond to conventional antibiotic regimens. A potential method to treat such chronic infections is to combine agents which interfere with bacterial biofilm development together with an antibiotic in a single formulation. Here, we explore the use of a new bioresponsive polymer formulation derived from specifically modified alginate nanoparticles (NPs) in order to deliver ciprofloxacin (CIP) in combination with the quorum sensing inhibitor (QSI) 3-amino-7-chloro-2-nonylquinazolin-4(3H)-one (ACNQ) to mature Pseudomonas aeruginosa biofilms. The alginate NPs were engineered to incorporate a pH-responsive linker between the polysaccharide backbone and the QSI, and to encapsulate CIP via charge–charge interactions of the positively-charged drug with the carboxyl residues of the alginate matrix. In this way, a dual-action release of antibiotic and QSI was designed for the low-pH regions of a biofilm, involving cleavage of the QSI-linker to the alginate matrix and reduced charge–charge interactions between CIP and the polysaccharide as the alginate carboxyl side-chains protonated. When tested in a biofilm model the concomitant release of CIP + QSI from the pH-responsive nanoparticles significantly reduced the viability of the biofilm compared with CIP treatment alone. In addition, the alginate NPs were shown to penetrate deeply into P. aeruginosa biofilms, which we attribute in part to the charges of the NPs and the release of the QSI agent. Finally, we tested the formulation in both a 2D keratinocyte and a 3D ex vivo skin infection model. The dual-action bio-responsive QSI and CIP release nanoparticles effectively cleared the infection in the latter, suggesting considerable promise for combination therapeutics which prevent biofilm formation as well as effectively killing mature P. aeruginosa biofilms.
Introduction
The increasing problems of multi-antibiotic resistance urgently require alternative therapeutic options.1–3 One of the main issues associated with indiscriminate antibiotic use is the activation of defence mechanisms in pathogens,4 and as a survival strategy, bacteria form biofilms.5 These are surface-associated multicellular communities embedded in a self-generated extracellular matrix (ECM).6 The latter is composed of polysaccharides, proteins and extracellular DNA (eDNA), and is both heterogeneous in composition and architecture, as well as resistant to penetration by antimicrobial agents.7 Upon administration, most antibiotics kill the more susceptible planktonic bacterial cells, leaving behind the intrinsically antibiotic tolerant biofilm embedded bacteria. The tolerance of biofilms to antimicrobial agents generally is multifactorial involving physical, physiological and intrinsic genetic determinants whereas antibiotic resistance is a consequence of mutations and the acquisition of antibiotic resistance genes via horizontal gene transfer.8 Biofilms are also refractory to clearance by the host immune system and so elicit a number of major clinical challenges, such as chronic inflammation, impaired wound healing and systemic spread of chronic infections.9 Novel therapeutic strategies are thus urgently needed to combat the threat of biofilm-centred infections.10–17One of the most promising approaches is the combined-use of antibiotics with adjuvants that do not affect the pathways essential for the bacterial growth and viability, and as a consequence are less likely to select for resistance. Such agents have various modes of action including (i) increasing bacterial cell membrane permeability, (ii) impairing biofilm formation, and/or the production of virulence factors and antibiotic resistance elements, (iii) blocking antibiotic efflux pumps and (iv) changing phenotype from the biofilm to the planktonic state through biofilm dispersal. Since bacterial communities co-ordinate their pathogenic activities including biofilm development through cell–cell communication (quorum sensing, QS)5,18 using small diffusible signal molecules, disruption of QS interferes with biofilm formation and maturation.19–23 However, while there are combination formulations in the clinic which use antibiotics and β-lactamase inhibitors (e.g. Tazocin®, Unasyn® Augmentin®), none to date have utilised QS inhibitors (QSIs) as biofilm disrupting agents. Several QS and QSI control and release systems have however been described, including molecularly imprinted polymers which bind the QS agents to suppress bacterial biofilm formation, and peptide mimetics which disrupt biofilms of several bacterial strains.24–29
Here we set out to prepare a new antimicrobial biofilm formulation which would have the following attributes, (i) QS inhibition, (ii) broad spectrum antibiotic activity, (iii) good biofilm penetration, and (iv) good tissue compatibility with low cytoxicity. We selected the clinically important bacterium Pseudomonas aeruginosa as the model organism, as this pathogen is responsible for more than 30% of hospital-acquired infections and causes chronic biofilm-centred infections in diverse body sites. This organism is also intrinsically resistant to many antibiotics and a member of the ESKAPE group of multi-antibiotic resistant bacterial pathogens.30 For the QS inhibitor (QSI), we selected 3-amino-7-chloro-2-nonylquinazolin-4(3H)-one (ACNQ), a potent inhibitor of the pseudomonas quinolone QS system which targets the PqsR receptor. ACNQ inhibits virulence factor production and biofilm development and in particular reduces the production of matrix components including eDNA and rhamnolipids.31 The second component was ciprofloxacin, (CIP), a fluoroquinolone antibiotic with broad spectrum activity but also a drug to which bacterial pathogens including P. aeruginosa have acquired resistance.32 In order to penetrate a dense biofilm matrix, we chose to encapsulate the antibiotic and conjugate the QSI within in situ-formed sodium alginate nanoparticles. These carry a net negative charge at the pH values likely to be encountered in many bacterial biofilms. In addition, alginate oligosaccharides have been reported to disrupt biofilm formation and maturation and potentiate antibiotic action against P. aeruginosa.33 We thus anticipated that the alginate nanoparticles might penetrate further into the biofilm since the ECM contains polyanion-containing exopolysaccharides and eDNA34 as well as the negatively charged P. aeruginosa cell envelope,35 compared with ACNQ (weakly basic) and CIP (zwitterionic, but with a net positive charge at pH 7.4). In addition, alginates are already widely used in oral formulations, have a proven safety profile, are readily available, and can be derivatised further due to their multiple carboxyl residues.36
The combined formulation thus consisted of alginate-based nanoparticles (NPs), containing CIP encapsulated by charge–charge interactions during a nanoprecipitation process, with ACNQ attached via a pH-responsive hydrazine linker, and of a size range (150–200 nm) similar to those of Pseudomonas bacteriophages.37 We then tested these formulations as synergistic adjuvant-drug therapeutics in infection models consisting of pre-established biofilms of P. aeruginosa PAO1-Nottingham subline (PAO1-N) of ≥100 μm thickness in vitro and in both 2D and 3D skin infection models. The formulation and anti-infective action is shown schematically in Fig. 1.
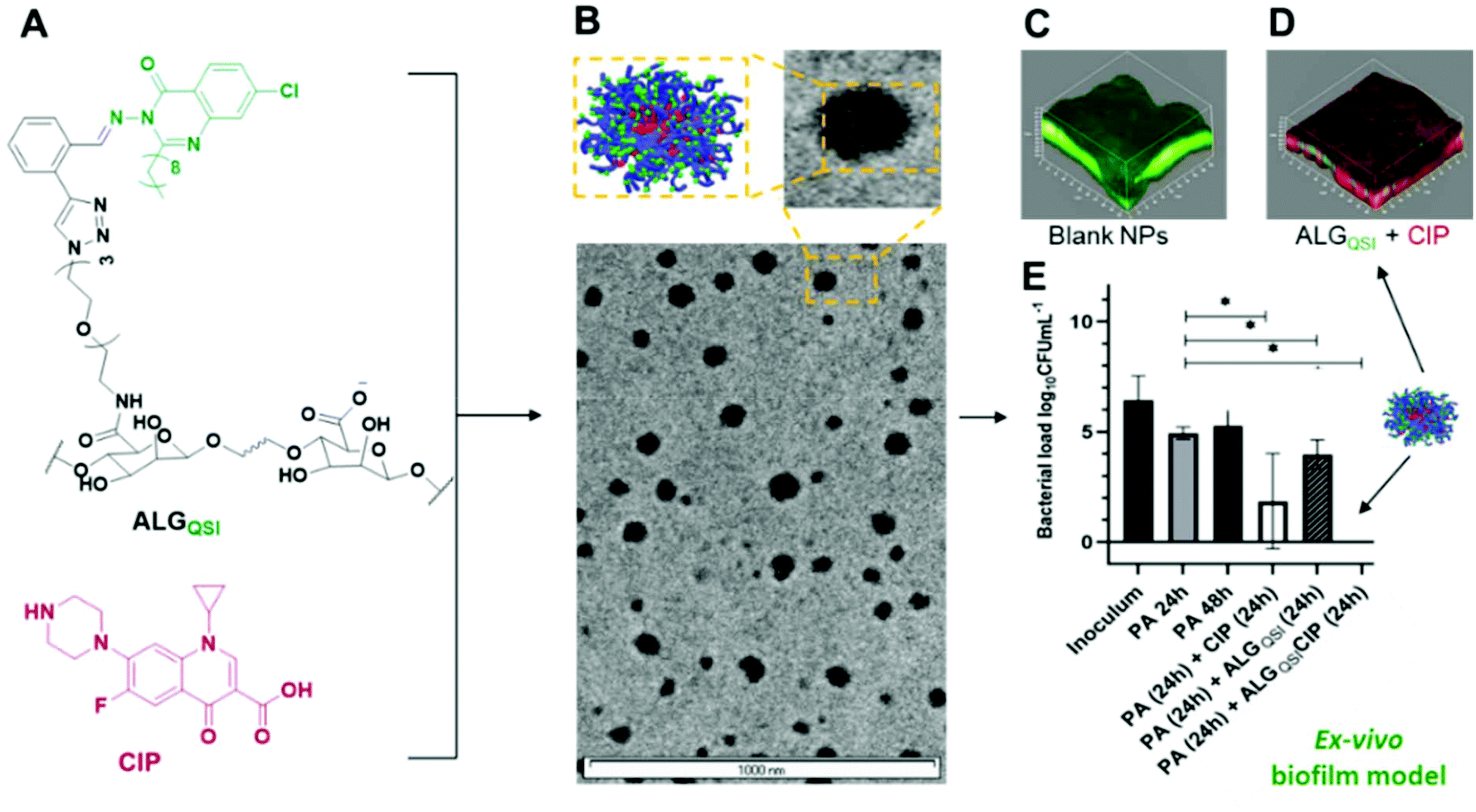 | ||
| Fig. 1 Schematic of combined polymer, QSI and antibiotic nanoparticle formulation (A, B) and activity against P. aeruginosa biofilms in vitro (C, D, E). Structures of components are shown in (A) with TEM micrographs in (B) In (C) viable P. aeruginosa are labelled green following treatment with blank NPs; while in (D) non-viable cells are labelled red following incubation with ALGQSI + CIP. Efficacy in an ex vivo infected skin model is shown in (E). | ||
Materials and methods
Experimental
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da, 3500 Da and 2000 Da MWCO) and Red fluorescent polystyrene-NH2 nanoparticles (λex ∼580 nm; λem ∼605 nm, d-200 nm) were purchased from ThermoFisher Scientific. All other analytical and synthesis grade solvents were purchased from ThermoFisher Scientific.
000 Da, 3500 Da and 2000 Da MWCO) and Red fluorescent polystyrene-NH2 nanoparticles (λex ∼580 nm; λem ∼605 nm, d-200 nm) were purchased from ThermoFisher Scientific. All other analytical and synthesis grade solvents were purchased from ThermoFisher Scientific.
Synthesis
1H NMR (CDCl3, 400 MHz, 25 °C), δ (ppm) 1H (9.55, s), 2H (8.26–8.24, d), 1H (7.72–7.73, d), 1H (7.67–7.62, dd), 2H, (7.60–7.50, p), 1H (7.44–7.41, dd), 1H (3.43, s), 2H (2.98–2.94, t), 2H (1.86–1.79, p), 2H (1.59, s), 2H (1.45–1.40, s), 8H (1.40–1.25, m), 3H (0.90–0.57, t). 13C NMR (101 MHz, CDCl3) δ 165.1, 158.24, 158.2, 147.4, 140.4, 134.3, 133.4, 131.9, 129.2, 128.8, 126.9, 126.7, 126.1, 124.4, 119.9, 84.0, 79.9, 35.2, 31.8, 29.4, 29.3, 29.3, 29.2, 26.7, 22.6, 14.1.
:8 mL – water:acetonitrile mixture at 55 °C. 11-Azido-3,6,9-trioxaundecan-1-amine (1 equiv., 2.08 mmol, 453 mg) was then added to the mixture along with the coupling reagents 2-chloro-4,6-dimethoxy-1,3,5-triazine (CDMT, 0.5 equiv., 1.04 mmol, 180 mg) and 4-methylmorpholine (NMM, 1 equiv., 2.08 mmol, 224 μl). The mixture was stirred at 55 °C for 16 h after which the solvent was removed under reduced pressure. The resulting solid was dissolved in water and filtered through cyano-modified silica gel to remove insoluble precipitates. The obtained solution was then dialyzed against a 10000 MWCO dialysis membrane for 2 days with DI water. The solution was then lyophilized to obtain the purified polymer. 1H NMR (D2O, 400 MHz, 25 °C), δ (ppm)-6.57 (s, Alg-CO–NH–CH2CH2–(OCH2CH2)3–N3), 5.10–4.90 (m, alginate), 4.60–3.80 (alginate, m), 3.80–3.50 (–(OCH2–CH2)3–O, 12H, m), 3.50–3.35 (Alg-CO–NH–CH2–CH2–O–, 2H, t), 3.20–3.10 (–OCH2–CH2–N3, 2H, t). IR (wavenumber, cm−1) – 1600 (CO–NH amide stretch), 2100 (N3 (azide) stretch), 2879, 2918 (CH stretch), 3278 (–COOH stretch).
:methanol 1:1 (5 mL) at 55 °C. To this solution were added tris[(1-benzyl 1H-1,2,3-triazol-4-yl) methyl]amine (TBTA, 0.2 equiv., 50 mg), triethylamine (0.25 equiv., typically 15 μl), and copper(I)-iodide (0.25 equiv., 5 mg) as coupling agents. Then 0.51 mmol of 2-ethynylbenzaldehyde (for ALGALD) or (Z)-7-chloro-3-((2-ethynylbenzylidene)amino)-2-nonylquinazolin-4(3H)-one (for ALGQSI) was added and the mixture was stirred at 55 °C overnight. The solvent was removed under reduced pressure and the oily product was resuspended in 5 mL of water and dialyzed. The resulting solution was then dialyzed using a 10000 MWCO dialysis membrane in phosphate buffer pH 7.4 to purify further the polymer, which was then recovered after lyophilisation.
ALGALD – 1H NMR (D2O, 400 MHz, 25 °C), δ (ppm) 9.88 (–CHO, 1H, s), 8.44 (triazole, 1H, s), 8.00–7.66 (aromatic from benzaldehyde, 4H, m), 7.47 (amide, 1H, m), 5.10–4.90 (alginate, 2H, m), 4.79 (H2O), 4.25–3.50 (alginate, m), 3.50–2.75 (Alg-CO–NH–CH2–CH2–(O–CH2–CH2)3–N-triazole) oliethyleneglycol (PEG3) (linker, m, 16H, m). 13C-(DMF, 120 MHz, 25 °C), δ (ppm)-195.4 (–CHO), 175.9 (–COOH-alginate), 175.4 (Alg-CONH-PEG3-), 149.9 (–N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–NN-triazole ring), 149.8 (–C–CHO-aromatic ring), 135, 134, 133, 130, 129.6 (aromatic Cs from benzaldehyde), 129.5 (N–CC–NN-triazole ring), 100.9, 100.6, 100.1, 99.8, (Alginate-C–O–C), 80.0, 77.2, 76.1, 72.0 (Alginate-C–OH), 68.5, 67.5 (Alg-CO–NH–CH2CH2–O–(CH2CH2)2–O–CH2–CH2-triazole), 67.0, 64.8, 62.4, 50.3, 46.6, 38.7 (Alg-CO–NH–CH2–CH2–(O–CH2CH2)2–O–CH2–CH2-triazole-). IR (wavenumber, cm−1) – 1600 (CONH amide carbonyl stretch), 1686 (–CHO carbonyl stretch), 2855 (aromatic CHO stretch), 2918 (triazole ring), 3062 (CH-alkane stretch from oligoethyl PEG3 linker), 3278 (–COOH stretch).
C–NN-triazole ring), 149.8 (–C–CHO-aromatic ring), 135, 134, 133, 130, 129.6 (aromatic Cs from benzaldehyde), 129.5 (N–CC–NN-triazole ring), 100.9, 100.6, 100.1, 99.8, (Alginate-C–O–C), 80.0, 77.2, 76.1, 72.0 (Alginate-C–OH), 68.5, 67.5 (Alg-CO–NH–CH2CH2–O–(CH2CH2)2–O–CH2–CH2-triazole), 67.0, 64.8, 62.4, 50.3, 46.6, 38.7 (Alg-CO–NH–CH2–CH2–(O–CH2CH2)2–O–CH2–CH2-triazole-). IR (wavenumber, cm−1) – 1600 (CONH amide carbonyl stretch), 1686 (–CHO carbonyl stretch), 2855 (aromatic CHO stretch), 2918 (triazole ring), 3062 (CH-alkane stretch from oligoethyl PEG3 linker), 3278 (–COOH stretch).
ALGQSI – 1H NMR (D2O/DMSO-d6-90/10, 400 MHz, 70 °C), δ (ppm)-8.60–7.40 (aromatic and hydrazone protons of ACNQ-triazole conjugate, m), 5.30–5.00 (alginate, 2H, m), 4.75–4.00 (alginate, m), 4.71 (HDO), 4.20, 3.90 and 3.60–3.90 (Alg-CONH–CH2–CH2–(O–CH2–CH2)3-triazole-, m), 2.50 (DMSO), 1.75–1.50 (m, –(CH2)9–CH3 alkyl chain of ACNQ). 13C-(DMF, 120 MHz, 25 °C), δ (ppm) 161.0 (Alg-CONH-PEG3-), 145.4 (–N–CO-ACNQ drug), 140.1 (–N–CC–NN-triazole ring), 137.7 (C9–C(–N–)–NN-ACNQ drug), 134.0 (tert-aromatic unsubstituted Cs of ACNQ QSI chlorinated ring), 129.8–129.1 (CC tert-aromatic), 127.3 (–N–NHC–, hydrazone bond), 125.3 (aromatic C–Cl, ACNQ QSI drug), 121.2 (quart-C–N, aromatic–Cl ring ACNQ), 74.8, 71.8, 71.6, 70 (–C–O–C, alginate), 64.9–64.8 (–C–CO–, alginate), 54.1 (Alginate-C–OH), 48.3, 46.5 (Alg-CO–NH–CH2CH2–O–(CH2CH2)2–O–CH2–CH2-triazole), 32.7–28.2–26.5 (Alg-CO–NH–CH2–CH2–(O–CH2CH2)2–O–CH2–CH2-triazole-), 23.5 (CH3–(CH2)7–CH2-ACNQ QSI drug), 14.7 (CH3–(CH2)7–CH2-ACNQ), 9.2 (CH3–(CH2)7–CH2-ACNQ).
IR (wavenumber, cm−1) 693, 713, 827 (C–Cl, heteroatom stretch from ACNQ), 1051, 1215 (C–N aliphatic stretch from ACNQ), 1510 (CN hydrazone stretch), 2918 (triazole ring), 3060 (CH-alkane stretch from alkyl chain of ACNQ and oligoethyl PEG3 linker), 3142 (aromatic stretch from ACNQ and aromtic linker), 3278 (–COOH stretch).
Determination of N3 functional groups in N3-TEG-ALG
The ERETIC method (Electronic REference To access In vivo Concentrations)38 was used in which 4 mg of N3-TEG-ALG was dissolved in 600 μL of D2O to which 0.275 mg of hydroquinone dissolved in 100 μL of D2O was added. The peak of hydroquinone at 6.7 ppm (4H) was used to calculate the concentration of N3-PEG3 groups in the N3-PEG3-ALG polymer.Protocol for nanoparticle (NP) formation
The NPs were prepared by a one-step in situ nanoprecipitation process. Briefly, the sodium alginate polymers (10 mg) were dissolved in PBS (10 mL) containing Tween 20 by sonication and added dropwise to a solution of CIP (10 mg) dissolved in 0.1 N HCl solution (0.1 mL). This solution was stirred overnight at RT. The solution obtained was dialysed against PBS for 72 hours to remove the free drug molecules. The dialysate was then freeze-dried and stored at 4 °C before use. The lyophilised NPs were equilibrated at room temperature and re-suspended in Tween 20 stabilised PBS (10 mL) before use. NPs without CIP were prepared by dissolving the 10 mg of polymer in 10 mL of 0.1 N HCl in buffer by sonication, followed by stirring overnight at RT.Morphology, particle size, and zeta potential of NPs
The morphologies of the NPs were characterized by transmission electron microscopy (TEM; Tecnai G2, FEI, Oregon USA). The samples for TEM were prepared by adding dropwise the NP solution (1 mg mL−1) onto the copper grid followed by drying for 4 hours before performing the microscopy. The particle sizes and zeta potentials of the NPs (1 mg mL−1) were determined using a Zetasizer Nano ZS90, Malvern Instruments Ltd, UK.Drug loading and pH-dependent release from the NPs
The amount of CIP in the NPs was quantified using a Cary 50 Bio UV spectrometer. Briefly, the amount of encapsulated CIP was obtained by dissolving different volumes of CIP-containing NPs in triplicate samples (1 μL, 2 μL, 3 μL, 4 μL, 5 μL) in 0.1 N HCl solution (∼1 mL) to release all the encapsulated CIP followed by dilution in PBS (total final volume-11 mL). The final concentration of CIP in the NPs was obtained (0.7 mg CIP per mL for ALGALDCIP and 0.8 mg CIP per mL for ALGQSICIP) by comparing the UV peak intensities with the calibration curve of CIP (λmax = 275 nm). The amount of ACNQ in the NPs was estimated by directly correlating the complete consumption of azide groups in N3-PEG3-ALG (following 2100 cm−1 peak corresponding to azide(N3) by IR) to the concentration of added QSI-alkyne molecule after the click reaction. The amount of ACNQ drug was considered the same as the amount of N3 groups previously present in ALG-PEG3-N3 as determined by the ERETIC NMR technique.For pH-responsive release, the release profiles of CIP (λmax = 275 nm) and ACNQ (λmax = 240 nm) from the NPs were studied using UV-VIS spectroscopy in pH 7.4 PBS buffer or pH 6.0 citric acid buffer. Typically, 2 mL of NPs solution (1.0 mg mL−1) were placed in a dialysis bag (MWCO = 10 kDa), which was then immersed in 5 mL of release medium at different pH values at 37 °C and with constant shaking at 100 rpm. At the predetermined time points, 3 mL of medium was collected and replaced by 3 mL of fresh buffer.
Bacterial growth inhibition
To evaluate the growth inhibitory activity of alginate-CIP conjugates, free or encapsulated CIP was tested against P. aeruginosa PAO1-N cultures at concentrations 0.01, 0.05, 0.1, 0.2, 0.5 and 1 μg mL−1. Bacteria were grown overnight in Luria broth (LB) at 37 °C. The next day the optical density (OD600 nm) of the culture was adjusted to 0.01 in FAB39 medium containing 10 mM glucose and 200 μL aliquots supplemented with CIP were loaded into the wells of a 96-well plate. As controls, medium only and wells containing NPs without ciprofloxacin were employed. Bacterial growth was monitored for 24 h at 37 °C in a 96-well plate TECAN Genios Pro spectrophotometer (Tecan, UK). The minimum inhibitory concentration (MIC) was defined as the antibiotic concentration where no visible bacterial growth was observed or OD600 nm was <10% compared with the untreated control after 24 h of antibiotic exposure.Biofilm viability and penetration assays
Mature pre-established PAO1-N biofilms were grown to assess the effects of co-delivered CIP and ACNQ on biofilm viability. For these assays, PAO1-N biofilms were grown on round glass coverslips (8 mm, #1.5 thickness) under flow conditions (20 rpm) in FAB containing 10 mM glucose medium inoculated with a diluted (OD600 nm = 0.01) bacterial suspension from overnight cultures in LB. Biofilms were cultivated at 30 °C for 3 days with medium replacement every day. Biofilms were washed in PBS to remove loosely attached cells, and incubated for a further 24 h in fresh FAB medium supplemented with various treatments. These included free CIP and/or ACNQ as well as the NPs: ALGAld (empty NPs), ALGQSI, ALGALDCIP and ALGQSICIP. With final concentrations of CIP and ACNQ of 60 μg mL−1 (300× MIC) and 4 μg mL−1 respectively. Biofilms exposed to each treatment were washed in PBS and the viabilities of the attached cells were evaluated using the LIVE/DEAD® BacLight™ Bacterial Viability kit (Molecular Probes, Life Technologies) according to the manufacturer's instructions. Following staining, coverslips were rinsed with sterile water and imaged using a LSM 700 laser scanning confocal microscope (CLSM) (Carl Zeiss, Germany). Viable and non-viable biomass quantification from image stacks of biofilms was done with Comstat2 software39 (http://www.comstat.dk). Live/dead ratios were established for each treatment and compared with untreated controls. Five independent assays were carried out in triplicate.To ascertain that the reduced viability of the biofilms exposed to combined therapy was retained at different levels of exposure, a dose–response validation was carried out using different concentrations of CIP (5, 20 and 60 μg mL−1 = 25×, 100× and 300× MIC respectively) while retaining the QSI concentration constant at 4 μg mL−1. The biofilm viability was compared with that of free CIP treatment and an untreated control.
To evaluate NP diffusion through the biofilm matrix, ALGALD and ALGQSI conjugates were labelled with fluorescein and tested against 3-day old PAO1-N biofilms. After 1, 2 and 24 h of incubation with NPs, biofilm samples were collected and stained with Syto64 fluorescent dye prior to CLSM image acquisition to simultaneously detect bacterial cells (red fluorescence) and NPs (green fluorescence). NP diffusion was determined by quantifying the fluorescein signal associated with each image stack and relating it to the biomass at different biofilm depths. Moreover, to relate biofilm penetration of alginate conjugates with other commercially available fluorescent NPs, the diffusion of blue fluorescent 50 nm polystyrene beads and red fluorescent 200 nm polystyrene beads carrying positive or negative zeta potentials were also tested in biofilms stained with Syto64 and Syto9 dyes respectively.
2D keratinocyte infection model
000 units)/streptomycin (10 mg mL−1) until the monolayer was 80–85% confluent. Cells were trypsinised using trypsin/EDTA and seeded at 45000 cells per cm2 in an 8 well μ-slide Ibitreat chamber (Ibidi, Germany). When 90–100% confluent, cells were washed three times with Dulbecco's phosphate-buffered saline (DPBS). Prior to infection, HaCat cells were stained using CellTracker Green CMFDA (Thermofisher), following the manufacturer's instructions.
000 and 150 μL were added to each well containing confluent HaCat cells. ALGQSI NPs containing 4 μg mL−1 of QSI or ALGALD (empty NPs) were added at a final concentration 300 μg mL−1. Infected cells were incubated at 37 °C, with 5% CO2 and 95% humidity for 20 h prior to imaging using CLSM.
3D skin infection model
Results and discussion
Polymer synthesis and characterisation
The design principles of the combination therapeutic were firstly that it needed to be derived from a simple, readily accessible polymer already used in human clinical applications. Secondly, the chemistries should enable a QSI to be attached via a pH-responsive linker and an antibiotic to be encapsulated. Finally, the whole system was required to self-assemble into lightly negatively-charged nanoparticles (NPs) in order to penetrate dense bacterial biofilms. Accordingly, we chose sodium alginates as the ‘platform’ material and modified the polymer with functionalities which enabled active agents to be incorporated via Schiff base linkages for in situ release in the acidic environment of a biofilm. We selected the QSI, 3-amino-7-chloro-2-nonylquinazolin-4(3H)-one (ACNQ) (Ilangovan et al. 2013)31,40, and utilised a triethyleneglycol spacer to append it to the alginate to ensure this hydrophobic drug was accessible to the aqueous environment when formulated. Finally, we opted for ciprofloxacin (CIP) as the antibiotic, as its positive charge at ambient pH values facilitated association with the alginate carboxylates and induced self-assembly of the system into NPs of sizes (150–200 nm) similar to those of Pseudomonas bacteriophages.37 The synthetic scheme for the construction of the dual bioresponsive combination therapeutic nanoparticles is shown in Scheme 1.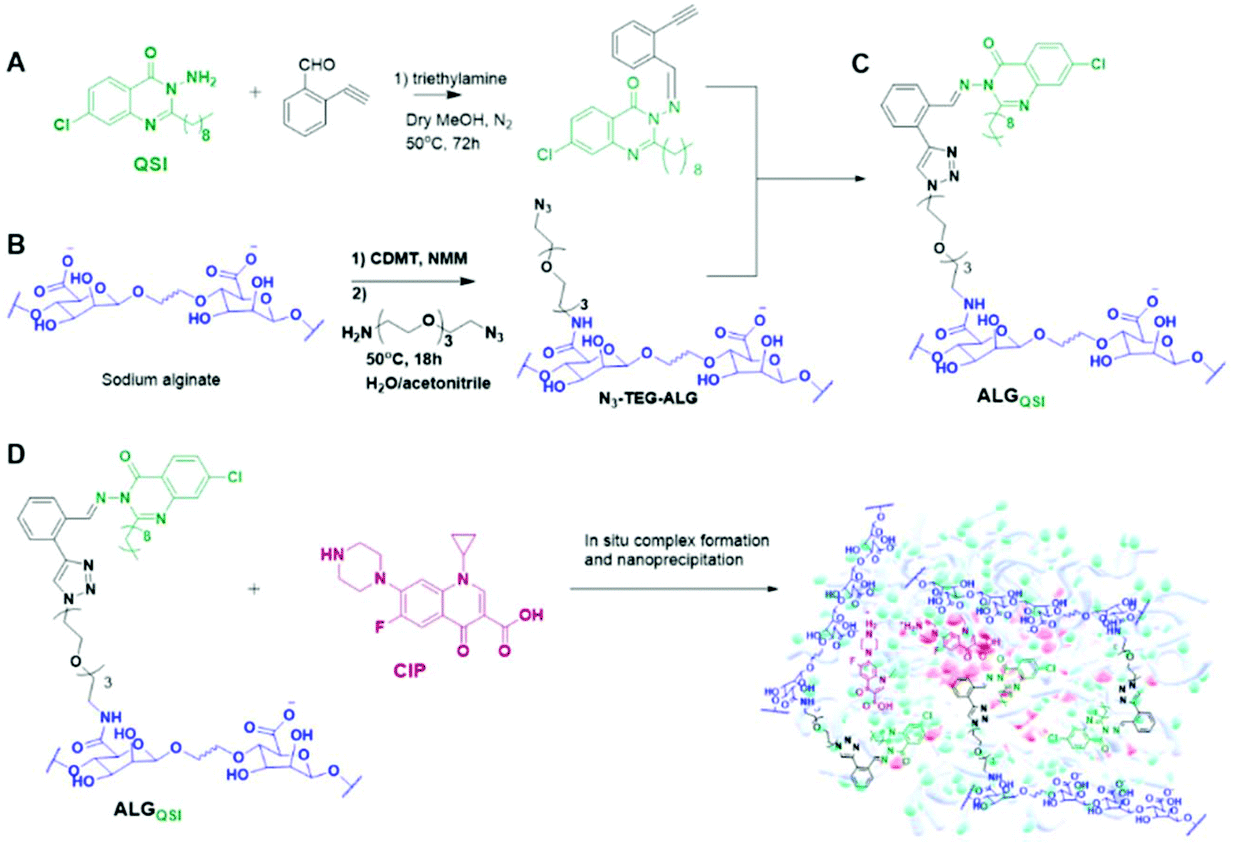 | ||
| Scheme 1 In (A) the QSI drug-3-amino-7-chloro-2-nonylquinazolin-4(3H)-one(ACNQ) was coupled with 2-ethynylbenzaldehyde to form the alkynyl-functional intermediate (Z)-7-chloro-3-((2-ethynylbenzylidene)amino)-2-nonylquinazolin-4(3H)-one. In (B) carboxyl groups in sodium alginate were first activated then reacted with an α,ω-amine and azide-functional triethyleneglycol to form N3-TEG-ALG, which was then ‘clicked’ (as shown in C) with (Z)-7-chloro-3-((2-ethynylbenzylidene)amino)-2-nonylquinazolin-4(3H)-one to form the ALGQSI polymer. In (D) Ciprofloxacin (CIP) was formulated into nanoparticles by an in situ nanoprecipitation process with ALGQSI. | ||
The synthesis started with Schiff base formation between 2-ethynylbenzaldehyde and ACNQ to form (Z)-7-chloro-3-((2-ethynylbenzylidene)amino)-2-nonylquinazolin-4(3H)-one in ∼80% yield. In parallel, CDMT/NMM based amide coupling was used to attach azide groups via a triethyleneglycol linker to the alginate backbone to obtain N3-TEG-ALG (Scheme 1). The derivatisation of the alginate backbone was confirmed by 1H NMR and IR, with the presence of the azide groups apparent in FTIR spectra at 2100 cm−1. The ERETIC method (Electronic REference To access In vivo Concentrations)38 was used to estimate the amount of free azide groups in N3-TEG-ALG (∼0.1 μmol/1 mg). For the next steps, the well-established Cu-catalysed azide–alkyne click reactions were used to attach (Z)-7-chloro-3-((2-ethynylbenzylidene)amino)-2-nonylquinazolin-4(3H)-one with N3-TEG-ALG to obtain the key functional polymer ALGQSI, (Scheme 1). We also prepared a second polymer, ALGALD, in which 2-ethynylbenzaldehyde was coupled to the N3-TEG-ALG backbone via the same click chemistry, but with no QSI attached. This was in order to render the polymer more amphiphilic, even without the QSI present, and thus potentially able to form nanoparticles with CIP in a manner analogous to that of ALGQSI polymers.
For both the derivatised alginates (ALGALD and ALGQSI), quantitative consumption of N3 groups upon completion of the click reaction was confirmed by complete disappearance on 2100 cm−1 peak in IR. In addition, FTIR signals at 1686 cm−1 and 2855 cm−1 for aromatic aldehyde carbonyl stretching, 2918 cm−1 for triazole, along with triazole + amide + aromatic signals (7.2–8.5 ppm), –CHO signal (10 ppm) in 1H NMR, and characteristic peaks in 13C NMR (for e.g. –CHO: 196 ppm) confirmed the successful formation of ALGALD. Similarly, for the ALGQSI polymer, characteristic IR absorption bands corresponding to CHNH stretching for hydrazones (1510 cm−1) and an intense band at 713 cm−1 for heteroatom C–Cl vibration confirmed the successful functionalisation with the QSI via a pH-responsive linkage. Characterisation of ALGQSIvia1H NMR was problematic at 25 °C due to the tendency for the polymers to self-assemble, leading to poorly-resolving signals. However, using elevated temperature NMR at 70 °C in a mixture of DMSO-d6 + D2O, the triazole + amide + aromatic signals in the range of 7.2–8.5 ppm, and ethylene glycol peaks (3.60–3.90 ppm) from N3-TEG-NH-were apparent alongside the alginate peaks. The successful coupling of the QSI on the N3-PEG3-ALG polymer was further verified by 13C NMR, and the absence of signals corresponding to –CHO in 1H, 13C and FTIR for ALGQSI also confirmed that the reversible hydrazone bond remained intact following synthesis and the conditions required for characterisation, with no evident liberation of ACNQ (Fig. 2).
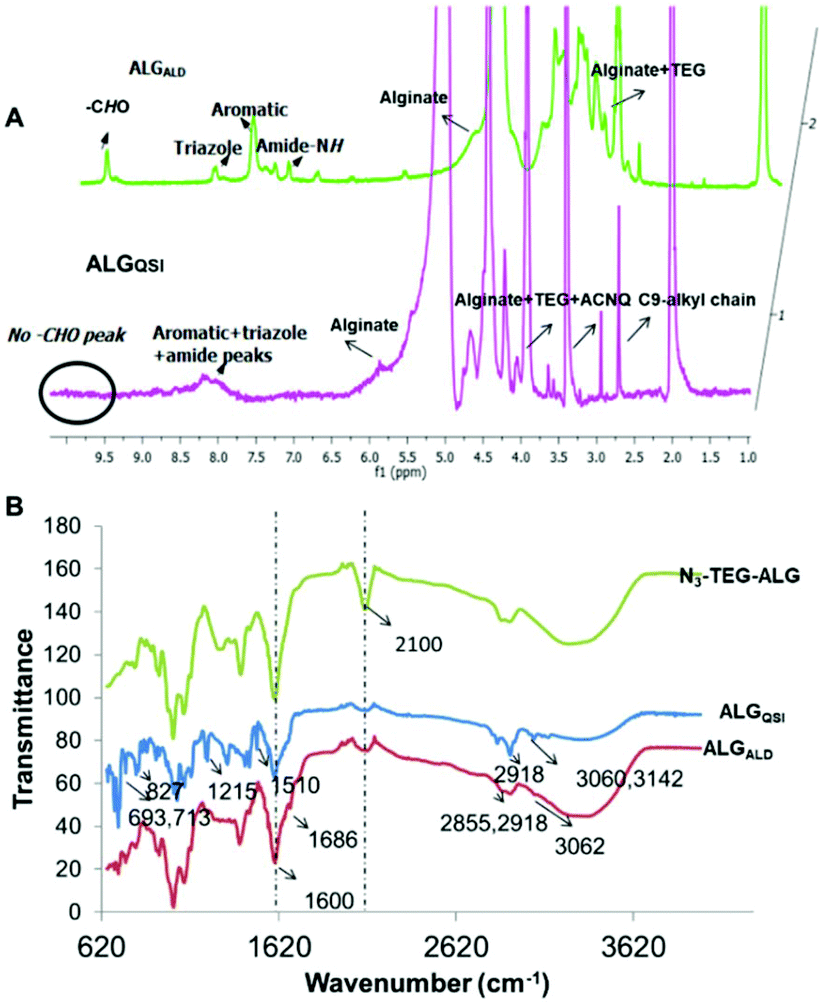 | ||
| Fig. 2 (A) 1H NMR spectra of ALGQSI at 70 °C and ALGALD at 25 °C. (B) FT-IR spectra comparing N3-PEG3-ALG, ALGQSI and ALGALD showing complete consumption of azide (2100 cm−1) for ALGQSI and ALGALD, and other characteristic peaks. | ||
Nanoparticle formulation and characterisation
After confirming the successful formation of the targeted amphiphilic structures-ALGALD and ALGQSI, the next step was to prepare self-assembled nanostructures. This was readily accomplished by a single step nanoprecipitation in phosphate buffer, owing to the amphiphilic character of the hydrophobic (triazole + ACNQ C9 chain in ALGQSI) and the hydrophilic (alginate and triethylene glycol) components. Both the polymers formed NPs with comparable diameters and zeta potentials (Table 1, Fig. 3A and B, Table S1, Fig. S11†). The negative zeta-potentials of the NPs with or without CIP confirmed that their outer exposed layer was predominantly the carboxyl-containing alginate chains.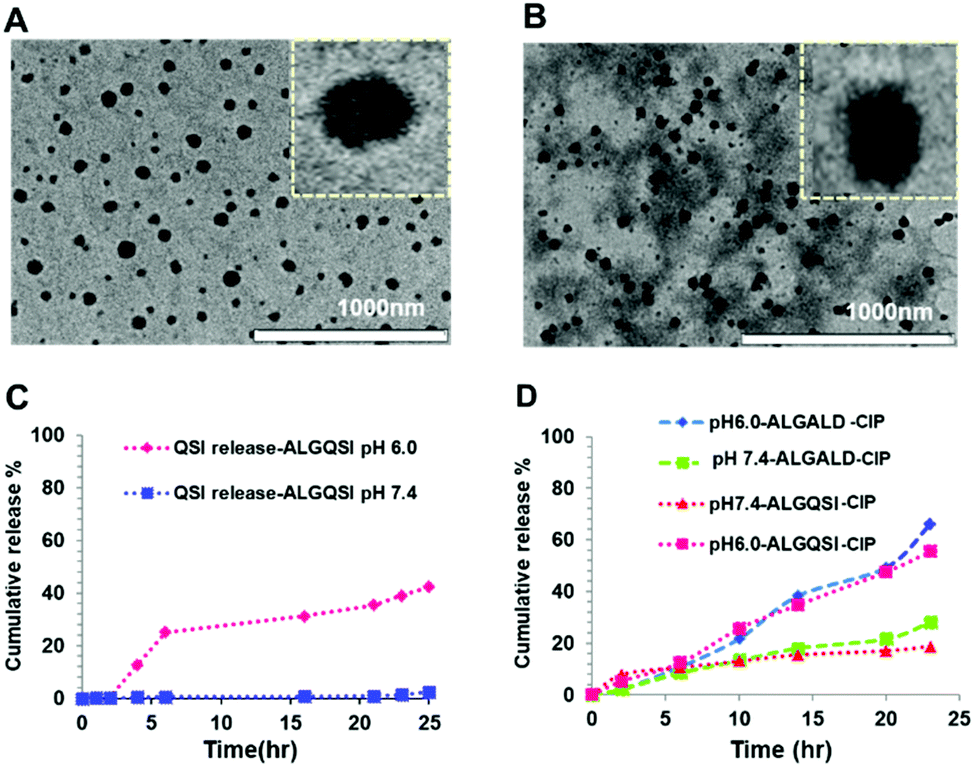 | ||
| Fig. 3 TEM images of (A) ALGQSICIP and (B) ALGALDCIP NPs. (C) Release profile over 24 h of ACNQ from ALGQSI at pH 6.0 and 7.4. (D) Ciprofloxacin release from ALGALD-CIP at pH 6.0 and 7.4. | ||
The in vitro drug release profiles from the NPs were evaluated at pH 7.4 and 6.0 that were considered likely to be encountered in ambient biological conditions and in an established biofilm, respectively. Higher cumulative release of the QSI drug from ALGQSI was observed at biofilm relevant pH 6.0 (0.5 M citrate buffer) than at pH 7.4 (0.1 M phosphate buffer), which was attributable to the accelerated hydrazone bond cleavage at the lower pH (Fig. 3C). Similarly, greater release of ciprofloxacin from both ALGQSI and ALGALD was observed at pH 6.0 than at pH 7.4 (Fig. 3D). This can be attributed to the high solubility of ciprofloxacin (pKa acid-6.16) at this pH range overcoming it's entrapment in the hydrophobic core along with the protonation of –COOH in alginate, weakening the electrostatic interaction between CIP and alginate.
In vitro efficacy studies
The biofilm ECM is a major barrier to drug transport and efficacy in biofilm killing and removal. Our hypothesis was that alginate-based nanoparticles would be advantageous in overcoming this barrier and distributing throughout the biofilm, especially as the net negative charges of the alginate-derivative NPs should prevent their accumulation at the biofilm surface. The latter problem limits the efficacy of many antimicrobial drugs, which are weakly basic and thus positively charged as they do not penetrate deeply into the biofilm core where antibiotic resistant or tolerant bacterial subpopulations are present.Dye-labelled alginates were prepared by coupling 6-amino fluorescein to the previously synthesised N3-TEG-ALG and the polymers were formulated as before into ∼200 nm nanoparticles. The NPs were administered to pre-established PAO1-N biofilms uniformly stained with SYTO64 (shown in blue in Fig. 4). No evident penetration by the fluorescent NPs was observed after 1 h incubation of the biofilms (fluorescein labelled NPs in yellow). However, after 2 h, the surface layers were partially covered with the NPs and within 24 h complete penetration throughout the biofilm was observed. In order to evaluate the role of alginate in determining the extent of biofilm penetration, we also tested commercially available fluorescent polystyrene NPs of comparable diameters (50 and 200 nm) with both +ve (commercially available) and −ve zeta potentials (derived by reaction of commercial –NH2 terminated polystyrene NPs with succinic acid). These NPs irrespective of their zeta potentials or diameters (50 nm–+53.7 mV and −32.6 mV, 200 nm–+48.4 mV and −32.5 mV) were unable to penetrate even after 24 h and stayed mostly at the outer surfaces of the biofilm (Fig. 4, ESI Fig. S16†). These assays confirmed the ability of the modified alginate NPs to penetrate the PAO1-N biofilms effectively and to a much greater extent than non-alginate-based NPs.
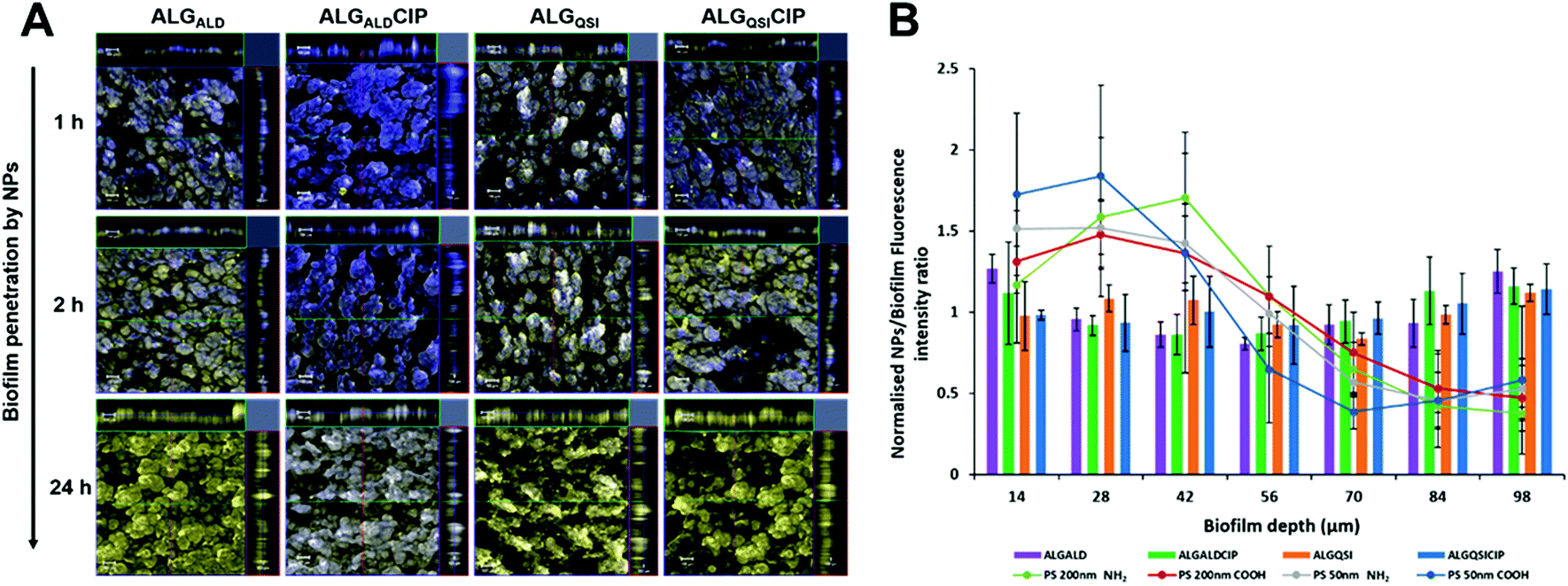 | ||
| Fig. 4 Penetration of P. aeruginosa biofilms by alginate nanoparticles. In (A), confocal laser scanning microscopy (CLSM) images show penetration of 6-amino fluorescein-ALGALD, ALGALDCIP, ALGQSI and ALGQSICIP NPs over time into PAO1-N biofilms stained with SYTO64 dye. The fluorescein labelled NPs are shown in yellow and the bacteria are stained blue. After incubating the biofilms for 1 h no penetration is observed as evident from the blue stained biofilm. The NPs begin to penetrate after 2 h of incubation as observed by the increase in yellow fluorescence from the biofilm structure. Within 24 h complete penetration to the biofilm bulk is observed, evident from the 3D Z-stack of the biofilm shown on the top and right of the main image. Scale bar: 100 μm. In (B), the penetration of the different ALG-based NPs within 24 h is quantified (bars), in contrast to the polystyrene (PS) NPs which are mostly present in the outer layer of the biofilm (28–42 μm depth, circles joined with lines). The fluorescence intensities of the ALG NPs and polystyrene NPs are normalised to the background fluorescence of the biofilm stain. | ||
To demonstrate the impact of combination therapy, we intended that ACNQ should act as a non-growth inhibitory adjuvant to CIP, by impacting on the biofilm architecture to render the biofilm sensitive to the killing action of the antibiotic. To validate this, we investigated the effects of ALGQSI NPs on the growth of planktonic PAO1-N for 24 h. The presence of NPs inflicted no adverse effect on bacterial growth. Similarly, the biofilm viability also remained unaltered upon administration of ALGQSI NPs over 24 h (ESI Fig. S13†). We also investigated whether the encapsulation of CIP in NPs could affect its activity by comparing the MICs of encapsulated and free CIP, which were demonstrated to be comparable (0.2 μg mL−1) (ESI Fig. S12 and S13†). After confirming that the adjuvant was not affecting the growth or viability of PAO1-N in planktonic or biofilm states, we evaluated the effect of CIP on biofilm viability when co-delivered with ACNQ in the alginate NPs.
PAO1-N biofilms were established for 3 days before challenging with NP-delivered therapeutics (Fig. 5) followed by Live/Dead staining. Untreated biofilms (Fig. 5A), those treated with buffer (Fig. 5B) or ACNQ only (Fig. 5C) showed only live bacteria. When the biofilms were exposed to CIP, some evidence of dead bacteria was obtained due to areas of red staining within the biofilm (Fig. 5D) and treatment with the quorum sensing inhibitor ACNQ combined with CIP resulted in enhanced cell kill, but in ‘patchy’ domains (Fig. 5E). The most effective treatment was with ALGQSI-CIP (Fig. 5F) in which deep penetration into the biofilm was apparent as evident from the large and diffuse red staining of dead bacteria.
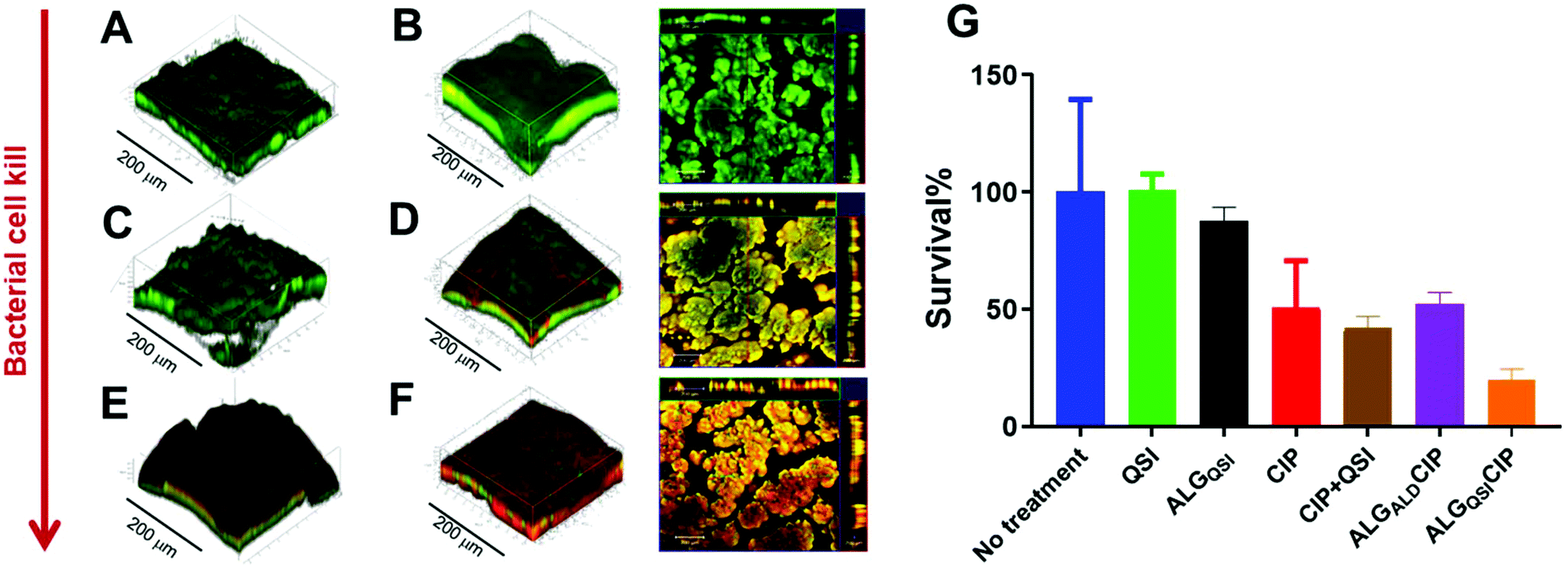 | ||
| Fig. 5 Representative CLSM 3D Z-stack images PAO1-N biofilms after 3 days’ growth and treatment for 24 hours with, (A) no drugs, (B) ALGQSI NPs, (C) ACNQ (4 μg mL−1) (D), CIP (60 μg mL−1) (E) a combination of QSI (4 μg mL−1) + CIP(60 μg mL−1), and (F) ALGQSICIP NPs with ACNQ(QSI)-4 μg mL−1 and CIP-60 μg mL−1. For images B, D and F, 2D microscopy images are included alongside the 3D CLSM, with overlapped live and dead imaged bacteria (depth profiles along the top and right borders). The live bacteria are depicted in green (SYTO9 dye) and the dead are shown in red colour (propidium iodide stain). In (G) are bar charts showing viability in PAO1-N biofilms quantified after treatment with different conditions for 24 hours. The concentrations of the drugs used were CIP-60 μg mL−1, CIP (60 μg mL−1) + ACNQ(QSI) (4 μg mL−1), ALGALDCIP (CIP-60 μg mL−1) and ALGQSICIP (ACNQ(QSI)-4 μg mL−1 + CIP-60 μg mL−1). | ||
Further quantification of the data is shown in Fig. 5G, including an additional formulation, ALGALD-CIP. These data showed that administration of CIP (60 μg mL−1 – i.e. 300 times the MIC of planktonic PAO1-N) to biofilms resulted in ∼50% cell kill after 24 h (Fig. 5G, red bar), and no noticeable improvement in the treatment of biofilm was observed by administering ACNQ in solution along with CIP (Fig. 5G, brown bar). In both the cases, the 3D images of biofilms post treatment revealed that only the outer surfaces of the biofilms exposed to the drugs were affected whereas the bulk of the biofilms remained unaltered (ESI Fig. S15 and S16†). This is consistent with low penetration of both drugs through the biofilm. Moreover, the formation of precipitate was observed in the growth medium after around 4 h of ACNQ administration. This was due to the extremely hydrophobic nature of ACNQ making its delivery impractical without a carrier system. The limited efficacy of CIP alone in these assays, and the poor solubility of ACNQ thus suggested that a suitable delivery system might better penetrate the biofilm enabling more effective release of drugs in situ. However, despite evidence of biofilm penetration by the alginate-based NPs, a formulation of CIP with the ALGALD NPs did not cause any further reduction in biofilm viability (Fig. 5G purple bar). However, the combined formulation, i.e. ALGQSICIP NPs containing both the antibiotic and ACNQ markedly reduced biofilm viability (Fig. 5F and G, orange bar). As evident from confocal microscopy, the bacterial viability was affected throughout the biofilm because of the deep penetration of alginate NPs and the likely delivery of both ACNQ and CIP in a spatio-temporally controlled manner. The viability of the biofilm was reduced to ∼19% of the original value by ALGQSICIP NPs compared to ∼50% for ALGALDCIP NPs applying the same dose of CIP (60 μg mL−1). Furthermore, the combined effect of CIP and ACNQ was apparent over a wide concentration range for ALGQSICIP NPs in terms of reduced biofilm viability, when compared with the corresponding concentrations of CIP alone (5–60 μg mL−1) (ESI Fig. S14†) Therefore, the alginate NPs mediated effective penetration and delivered a QSI-induced increase in biofilm susceptibility to antibiotic.
2D keratinocyte infection model
In addition to the effectiveness of the NP formulation in killing a preformed P. aeruginosa biofilm, its ability to prevent biofilm formation was also tested in a 2D keratinocyte skin epidermal cell infection model. Both the empty (ALGALD) and the NPs containing the QSI alone (ALGQSI) were added at the time of bacterial inoculation and biofilm formation was assessed after 20 h. Fig. 6 shows that the presence of ALGQSI significantly reduced biofilm formation (p-value <0.0001). However, it was apparent that the addition of the empty NPs alone also exerted a negative impact on biofilm formation (p-value <0.0001 for the 3 parameters) in this model. When comparing the impact of ALGQSI and ALGAL NPS on biofilm formation, bio-volume and average thickness but not surface area were significantly reduced in the presence of the QSI (p-values of 0.0105 and 0.0036 respectively; Fig. 6). Importantly, administration of ALGQSI NPs protected the HaCat cells to a much greater extent than the untreated layer or those or treated with the ALGALD NPs (Fig. 6).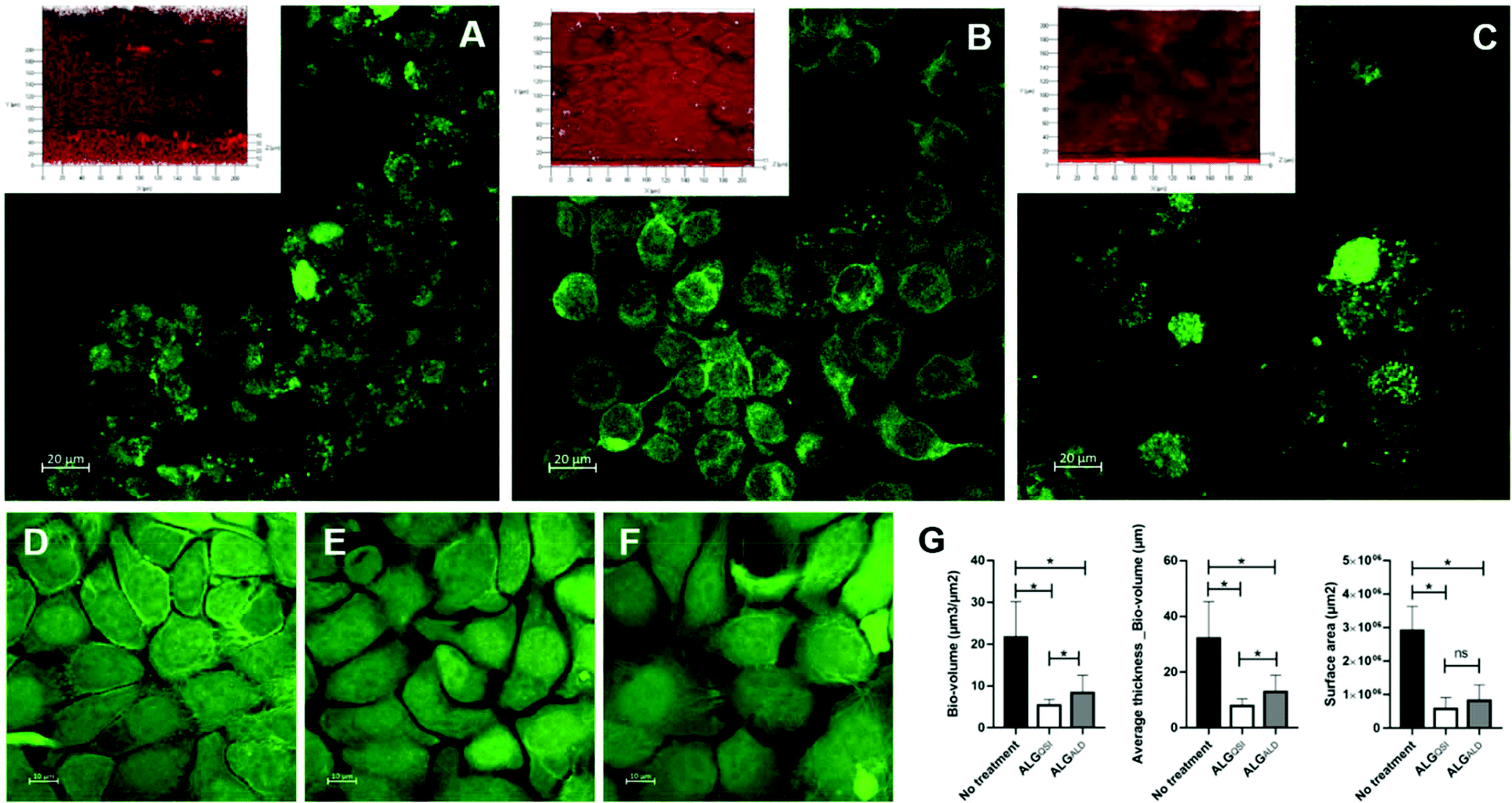 | ||
| Fig. 6 2D Keratinocyte Infection Model. HaCat cell monolayer infected with PAO1-N for 20 h (A) and treated with ALGQSI (B) or ALGALD (C) NPs (Image inserts show representative PAO1-N biofilms obtained). D, E & F: CLSM images of uninfected HaCat cells untreated (D), uninfected with 20 h ALGQSI treatment (E) and uninfected with 20 h ALGALD treatment (F). In G, bar charts showing (L–R) the effects of the different treatments on PA biofilm volume, average thickness and surface area. *: Statistically significant (p-value <0.05). The figure represents the average of four repeats. | ||
3D skin infection model
The efficacy of the NPs on a pre-existing P. aeruginosa skin infection was also tested. In this assay, ex vivo porcine skin plugs were inoculated with P. aeruginosa and after 24 h of infection, NPs were introduced and the skin incubated for a further 24 h. The viable bacterial loads recovered from the skin plugs were determined and the data are depicted in Fig. 7.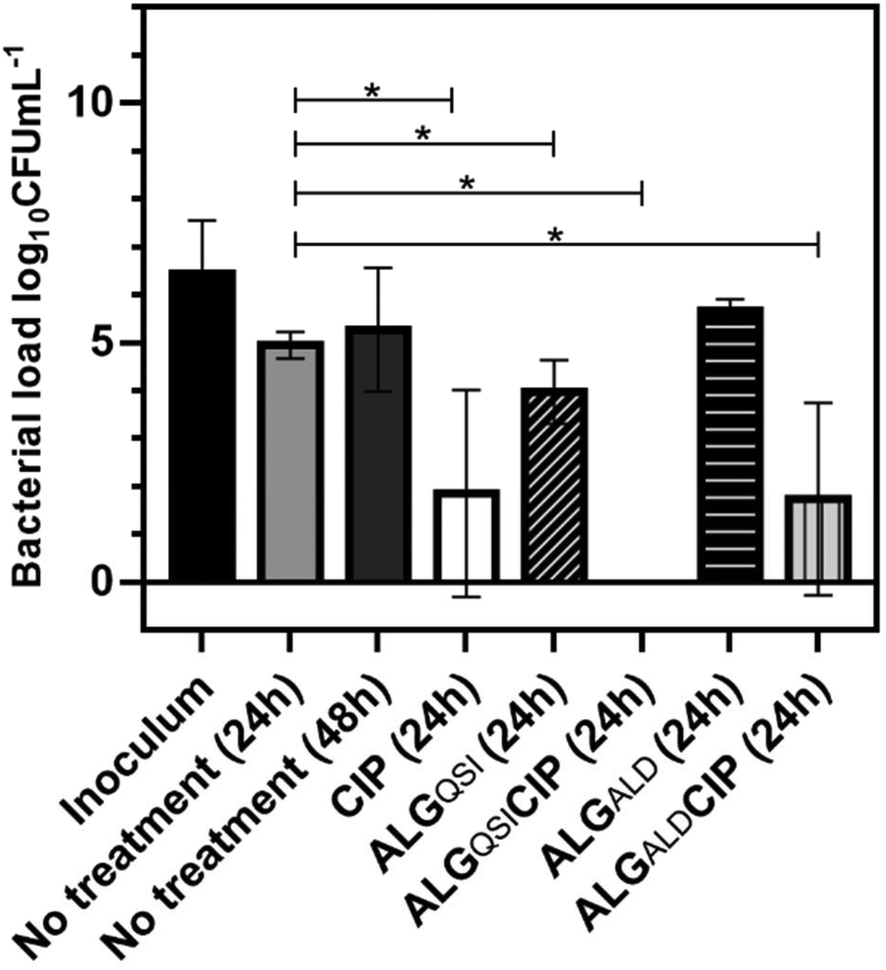 | ||
| Fig. 7 Bacterial load (log10CFU mL−1) recovered from skin infected with P. aeruginosa (PA) after 24 h or 48 h of incubation and/or after 24 h treatment with ciprofloxacin (CIP), ALGQSI, ALGQSICIP, ALGALD, ALGALDCIP. The bacterial inoculum is also represented. *: Statistically significant (p-value < 0.05). The figure depicts the average of 4 experiments. | ||
It is apparent from Fig. 7 that CIP alone (p-value <0.0001), ALGALDCIP (p-value <0.0001) and ALGQSI (p-value = 0.0048) each significantly reduced the bacterial load. However, the combination of the QSI and CIP, when included in the alginate nanoparticle formulation, completely cleared the infection (p-value <0.0015).
These data together show that the combination NP therapeutic was the most effective against the microbial communities in both 2D and 3D assays and, most importantly, in a relevant ex vivo infected skin model. It is well established that bacterial-biofilm centred infections are recalcitrant, chronic and difficult to eradicate with conventional antibiotic therapy,8 and that new treatment modalities are urgently needed. P. aeruginosa biofilms are frequently associated with recurrent respiratory tract infections in cystic fibrosis and chronic obstructive pulmonary disease as well as diabetic wounds and implanted medical device (e.g. catheter) associated infections. Biofilms in general are highly tolerant to antibiotics and often require concentrations over 1000 times higher than those required to kill planktonic cells although newly formed biofilms are more susceptible than mature biofilms.8 Therefore, the need for more effective treatments capable of killing established biofilms is also paralleled by the requirement for methods to prevent biofilm formation. Here we have shown that alginate NPs are not only capable of penetrating and delivering an antimicrobial agent payload deep into a P. aeruginosa biofilm but are also, in the absence of a payload, able to reduce biofilm formation in an infection model where the biofilm is allowed to form on a keratinocyte skin cell monolayer. However, the inclusion of a biofilm disrupting QSI, not only reduced biofilm formation to a greater extent but also provided some protection for the keratinocytes. In a more sophisticated ex vivo porcine skin model, the incorporation of both an antibiotic and a QSI within the alginate NP delivery system was required to eradicate completely the viable bacteria. As observed for the pre-formed biofilm, the dual action NP delivery system was far more effective than ciprofloxacin in solution, suggesting a clear advantage of incorporating the antibiotic in a carrier. In addition, the combined delivery system effectively killed the biofilm in situ with little apparent dispersal of the biofilm bacteria. This may offer an additional advantage in clinical practice, in that it reduces the likelihood of bacterial aggregates breaking away to initiate biofilm formation at new sites. Furthermore, while the QSI ACNQ has previously been shown to inhibit biofilm formation,40 its effect on preformed biofilms when delivered in NPs had not been shown before. Although ACNQ has a micromolar IC50, more potent PqsR analogues with nanomolar efficacies have recently been described and may offer improved potency as antibiotic adjuvants when delivered in NPs.23,41 Alternatively, a more rapid route to clinical testing could be achieved by incorporating re-purposed FDA-approved drugs such as clofoctol, an compound that acts as an antibiotic against Gram-positive but not Gram-negative bacteria but is a potent PqsR antagonist for P. aeruginosa.42
For the delivery system we selected alginate as the carrier polymer, in part because alginate oligomers have previously been shown to inhibit biofilm formation and potentiate the action of antibiotics against P. aeruginosa.33,43 However, P. aeruginosa strains produce heterogeneous ECMs that may contain one or more of three different exopolysaccharides including alginate.44 The P. aeruginosa strain PAO1-N used in our assays do not produce alginate containing biofilms and therefore it would be of interest to determine the efficacy of the NP delivery system in future against the mucoid-alginate producing P. aeruginosa strains that are frequently found in the lungs of individuals with cystic fibrosis who are chronically colonized by P. aeruginosa.43,44 In addition, further work will be required to determine whether dual action NPs are capable of eradicating P. aeruginosa biofilms in murine infection models and can be formulated for the treatment and prevention and treatment of chronic wounds and medical device associated infections. The advantages of a nanoparticulate formulation in this setting are the ability to incorporate multiple agents which could in principle be released by a range of in situ disease-related triggers, such as inflammatory oxidative stress45 in addition to local pH, thus enabling tuning of therapy to particular wound conditions. Finally, it would be of interest to evaluate the effects of functionality, charge, polymer/nanoparticle size and architecture systematically in bacterial recognition, biofilm penetration and therapeutic delivery.46–49 It is well-established that natural nanoparticulates such as viruses can penetrate through many biological barriers, and biomimicry of bacteriophage structural features might in future allow development of highly active synthetic NPs which exhibit functional mimicry and anti-bacterial action also.
Conclusions
In this study we have shown that alginate biopolymers can be modified to contain a responsive linker to the QSI ACNQ, and induced to self-assemble in the presence of an antibiotic, ciprofloxacin, to generate well-defined nanoparticles. These materials were able to release both the encapsulated antibiotic and the conjugated QSI in response to pH values expected in bacterial biofilms. When evaluated for efficacy against P. aeruginosa biofilms the nanoparticles were more effective in bacterial killing than the antibiotic alone, and the extent of cell killing was retained deep into the biofilm layer. The most significant data related to the activity of the nanoparticles in 2D keratinocyte and 3D ex vivo skin infection models. The alginate NPs showed some protective activity against P. aeruginosa infection in keratinocytes even in the absence of antibiotic, but the full combination therapeutic system completely cleared the infection in the 3D ex vivo skin model. In summary, the results demonstrate considerable promise for nanoparticle combination therapeutics, in the prevention of biofilm formation, in the reduction in biofilm load and through enhanced cell killing in established biofilms of the important ESKAPE pathogen P. aeruginosa.Data access statement
All raw data created during this research are openly available from the corresponding author (cameron.alexander@nottingham.ac.uk) and at the University of Nottingham Research Data Management Repository (https://rdmc.nottingham.ac.uk/) and all analysed data supporting this study are provided in the ESI† accompanying this paper.Conflicts of interest
There are no conflicts to declare.Funding sources
This work was supported by the Engineering and Physical Sciences Research Council [grant numbers EP/N006615/1 and EP/K005138/1, EP/N03371X/1]; the Biotechnology and Biological Sciences Research Council (BBSRC) [grant number BB/R012415/1] and the Royal Society [Wolfson Research Merit Award WM150086] (to CA). This project has received funding from the EMPIR programme co-financed by the Participating States and from the European Union's Horizon 2020 research and innovation programme [grant reference 15HLT01 MetVBadBugs]Acknowledgements
We thank the Nanoscale and Microscale Research Centre and the School of Life Sciences Imaging Facility (SLIM) at the University of Nottingham for providing access to advanced light and scanning microscopy instrumentation, and Drs Emily Smith and Tim Self at the University of Nottingham for valuable guidance. We also thank Christine Grainger-Boultby, Esme Ireson and Paul Cooling for skilled technical assistance, Alex Truman for ACNQ synthesis and Dr Sian Rankin-Turner, Elizabeth Hufton and Carol Turrill for invaluable project support.Notes and references
- B. D. Brooks and A. E. Brooks, Adv. Drug Delivery Rev., 2014, 78, 14–27 CrossRef CAS PubMed.
- M. Lakemeyer, W. N. Zhao, F. A. Mandl, P. Hammann and S. A. Sieber, Angew. Chem., Int. Ed., 2018, 57, 14440–14475 CrossRef CAS PubMed.
- P. Speck, Nature, 2013, 496, 169 CrossRef CAS PubMed.
- S. R. Partridge, S. M. Kwong, N. Firth and S. O. Jensen, Clin. Microbiol. Rev., 2018, 31(4), e00088-17 CrossRef PubMed.
- S. Mukherjee and B. L. Bassler, Nat. Rev. Microbiol., 2019, 17, 371–382 CrossRef CAS PubMed.
- H. Koo, R. N. Allan, R. P. Howlin, P. Stoodley and L. Hall-Stoodley, Nat. Rev. Microbiol., 2017, 15, 740–755 CrossRef CAS PubMed.
- R. M. Donlan and J. W. Costerton, Clin. Microbiol. Rev., 2002, 15, 167–193 CrossRef CAS PubMed.
- O. Ciofu and T. Tolker-Nielsen, Front. Microbiol., 2019, 10, 913 CrossRef PubMed.
- L. Hall-Stoodley, J. W. Costerton and P. Stoodley, Nat. Rev. Microbiol., 2004, 2, 95–108 CrossRef CAS PubMed.
- T. Parandhaman and S. K. Das, Biomater. Sci., 2018, 6, 3356–3372 RSC.
- N. Perez-Soto, L. Moule, D. N. Crisan, I. Insua, L. M. Taylor-Smith, K. Voelz, F. Fernandez-Trillo and A. M. Krachler, Chem. Sci., 2017, 8, 5291–5298 RSC.
- N. Doroshenko, S. Rimmer, R. Hoskins, P. Garg, T. Swift, H. L. M. Spencer, R. M. Lord, M. Katsikogianni, D. Pownall, S. MacNeil, C. W. I. Douglas and J. Shepherd, Biomater. Sci., 2018, 6, 2101–2109 RSC.
- L. T. Liu, B. Ercan, L. L. Sun, K. S. Ziemer and T. J. Webster, ACS Biomater. Sci. Eng., 2016, 2, 122–130 CrossRef CAS.
- C. F. Garcia, F. Stangl, A. Gotz, W. N. Zhao, S. A. Sieber, M. Opitz and O. Lieleg, Biomater. Sci., 2019, 7, 220–232 RSC.
- J. X. Guo, Y. Liu, Y. L. Chen, J. Q. Li and H. X. Ju, Chem. Sci., 2018, 9, 5906–5911 RSC.
- I. Di Bonaventura, X. Jin, R. Visini, D. Probst, S. Javor, B. H. Gan, G. Michaud, A. Natalello, S. M. Doglia, T. Kohler, C. van Delden, A. Stocker, T. Darbre and J. L. Reymond, Chem. Sci., 2017, 8, 6784–6798 RSC.
- B. Wang, Z. Ye, Q. Xu, H. Liu, Q. Lin, H. Chen and K. Nan, Biomater. Sci., 2016, 4, 1731–1741 RSC.
- J. S. Dickschat, Nat. Prod. Rep., 2010, 27, 343–369 RSC.
- K. Reuter, A. Steinbach and V. Helms, Perspect. Med. Chem., 2016, 8, 1–15 Search PubMed.
- P. Williams, K. Winzer, W. C. Chan and M. Camara, Philos. Trans. R. Soc., B, 2007, 362, 1119–1134 CrossRef CAS PubMed.
- B. Rémy, S. Mion, L. Plener, M. Elias, E. Chabrière and D. Daudé, Front. Pharmacol., 2018, 9, 203–203 CrossRef PubMed.
- P. Williams, Emerging Top. Life Sci., 2017, 1, 23–30 CrossRef.
- F. Soukarieh, P. Williams, M. J. Stocks and M. Cámara, J. Med. Chem., 2018, 61, 10385–10402 CrossRef CAS PubMed.
- M. J. Kratochvil, Y. Tal-Gan, T. Yang, H. E. Blackwell and D. M. Lynn, ACS Biomater. Sci. Eng., 2015, 1, 1039–1049 CrossRef CAS PubMed.
- E. Cavaleiro, A. S. Duarte, A. C. Esteves, A. Correia, M. J. Whitcombe, E. V. Piletska, S. A. Piletsky and I. Chianella, Macromol. Biosci., 2015, 15, 647–656 CrossRef CAS PubMed.
- E. V. Piletska, G. Stavroulakis, L. D. Larcombe, M. J. Whitcombe, A. Sharma, S. Primrose, G. K. Robinson and S. A. Piletsky, Biomacromolecules, 2011, 12, 1067–1071 CrossRef CAS PubMed.
- J. K. Vasquez and H. E. Blackwell, ACS Infect. Dis., 2019, 5, 484–492 CrossRef CAS PubMed.
- M. E. Boursier, J. B. Combs and H. E. Blackwell, ACS Chem. Biol., 2019, 14, 186–191 CrossRef CAS PubMed.
- A. G. Palmer, A. C. Senechal, T. C. Haire, N. P. Mehta, S. D. Valiquette and H. E. Blackwell, ACS Chem. Biol., 2018, 13, 3115–3122 CrossRef CAS PubMed.
- J. N. Pendleton, S. P. Gorman and B. F. Gilmore, Expert Rev. Anti-Infect. Ther., 2013, 11, 297–308 CrossRef CAS PubMed.
- A. Ilangovan, M. Fletcher, G. Rampioni, C. Pustelny, K. Rumbaugh, S. Heeb, M. Camara, A. Truman, S. R. Chhabra, J. Emsley and P. Williams, PLoS Pathog., 2013, 9(7), e1003508 CrossRef CAS PubMed.
- T. F. Mah, B. Pitts, B. Pellock, G. C. Walker, P. S. Stewart and G. A. O'toole, Nature, 2003, 426, 306–310 CrossRef CAS PubMed.
- S. Khan, A. Tondervik, H. Sletta, G. Klinkenberg, C. Emanuel, E. Onsoyen, R. Myrvold, R. A. Howe, T. R. Walsh, K. E. Hill and D. W. Thomas, Antimicrob. Agents Chemother., 2012, 56, 5134–5141 CrossRef CAS PubMed.
- C. Berne, A. Ducret, G. G. Hardy and Y. V. Brun, Microbiol. Spectrum, 2015, 3(4), MB-0018-2015 Search PubMed.
- J. Shephard, A. J. McQuillan and P. J. Bremer, Appl. Environ. Microbiol., 2008, 74, 6980–6986 CrossRef CAS PubMed.
- S. M. Morgan, A. Alshamkhani, D. Callant, E. Schacht, J. F. Woodley and R. Duncan, Int. J. Pharm., 1995, 122, 121–128 CrossRef CAS.
- X. Cui, J. You, L. Sun, X. Yang, T. Zhang, K. Huang, X. Pan, F. Zhang, Y. He and H. Yang, Sci. Rep., 2016, 6, 39130 CrossRef CAS PubMed.
- S. Akoka, L. Barantin and M. Trierweiler, Anal. Chem., 1999, 71, 2554–2557 CrossRef CAS PubMed.
- A. Heydorn, A. T. Nielsen, M. Hentzer, C. Sternberg, M. Givskov, B. K. Ersboll and S. Molin, Microbiology, 2000, 146(Pt 10), 2395–2407 CrossRef CAS PubMed.
- A. Ilangovan, M. Fletcher, G. Rampioni, C. Pustelny, K. Rumbaugh, S. Heeb, M. Cámara, A. Truman, S. R. Chhabra, J. Emsley and P. Williams, PLoS Pathog., 2013, 9, e1003508 CrossRef CAS PubMed.
- F. Soukarieh, E. Vico Oton, J. F. Dubern, J. Gomes, N. Halliday, M. de Pilar Crespo, J. Ramirez-Prada, B. Insuasty, R. Abonia, J. Quiroga, S. Heeb, P. Williams, M. J. Stocks and M. Camara, Molecules, 2018, 23, 257 CrossRef PubMed.
- F. D'Angelo, V. Baldelli, N. Halliday, P. Pantalone, F. Polticelli, E. Fiscarelli, P. Williams, P. Visca, L. Leoni and G. Rampioni, Antimicrob. Agents Chemother., 2018, 62(11), e01296-18 CrossRef PubMed.
- L. C. Powell, M. F. Pritchard, E. L. Ferguson, K. A. Powell, S. U. Patel, P. D. Rye, S.-M. Sakellakou, N. J. Buurma, C. D. Brilliant, J. M. Copping, G. E. Menzies, P. D. Lewis, K. E. Hill and D. W. Thomas, npj Biofilms Microbiomes, 2018, 4, 13 CrossRef PubMed.
- D. J. Wozniak, T. J. Wyckoff, M. Starkey, R. Keyser, P. Azadi, G. A. O'Toole and M. R. Parsek, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 7907–7912 CrossRef CAS PubMed.
- G. Saravanakumar, J. Kim and W. J. Kim, Adv. Sci., 2017, 4, 1600124 CrossRef PubMed.
- E. Leire, S. P. Amaral, I. Louzao, K. Winzer, C. Alexander, E. Fernandez-Megia and F. Fernandez-Trillo, Biomater. Sci., 2016, 4, 998–1006 RSC.
- N. Perez-Soto, O. Creese, F. Fernandez-Trillo and A. M. Krachler, ACS Chem. Biol., 2018, 13, 3021–3029 CrossRef CAS PubMed.
- E. P. Magennis, F. Fernandez-Trillo, C. Sui, S. G. Spain, D. J. Bradshaw, D. Churchley, G. Mantovani, K. Winzer and C. Alexander, Nat. Mater., 2014, 13, 748–755 CrossRef CAS PubMed.
- L. T. Lui, X. Xue, C. Sui, A. Brown, D. I. Pritchard, N. Halliday, K. Winzer, S. M. Howdle, F. Fernández Trillo, N. Krasnogor and C. Alexander, Nat. Chem., 2013, 5, 1058–1065 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9bm00773c |
| ‡ These authors contributed equally to this work. |
| § Current affiliation: Antibiotic Resistance and Pathogenicity of Bacterial Infections Group, Balearic Islands Health Research Institute (IdISBa), 07120, Palma, Spain. |
| This journal is © The Royal Society of Chemistry 2019 |