
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancement of siRNA transfection by the optimization of fatty acid length and histidine content in the CPP†
Ly
Porosk
 *a,
Piret
Arukuusk
a,
Kaisa
Põhako
a,
Kaido
Kurrikoff
a,
Kristina
Kiisholts
a,
Kärt
Padari
b,
Margus
Pooga
a and
Ülo
Langel
ac
*a,
Piret
Arukuusk
a,
Kaisa
Põhako
a,
Kaido
Kurrikoff
a,
Kristina
Kiisholts
a,
Kärt
Padari
b,
Margus
Pooga
a and
Ülo
Langel
ac
aInstitute of Technology, University of Tartu, Tartu, Estonia. E-mail: ly.porosk@ut.ee; Tel: +372737 4871
bInstitute of Molecular and Cell Biology, University of Tartu, Tartu, Estonia
cDepartment of Biochemistry and Biophysics, Stockholm University, Sweden
First published on 14th August 2019
Abstract
Extracellular synthetic nucleic acids, such as siRNAs, are unable to reach their intended targets efficiently. Therefore, delivery methods such as cell-penetrating peptides (CPP), which increase their transport, could enhance the potency of siRNA as therapeutic agents. The CPP NickFect55 (NF55) is an efficient peptide-based delivery vector, which has been previously used to deliver plasmid DNA into cells in vivo. To achieve higher intracellular delivery and bioactivity from the delivered cargo, we designed a series of histidine-containing peptides by optimizing pH-sensitivity, net charge, hydrophobicity, and charge distribution in the sequence of the CPP NF55. In the current work, we have applied a strategy where we have replaced amino acids in the C-terminus of the peptide in order to distribute hydrophobic and hydrophilic amino acids into distinct regions along the alpha-helical projection, including histidine amino acids into the sequence at the N-terminus, and optimizing the N-terminal fatty acid modification to suit the specific peptide sequence. We tested the CPPs based on the transfection efficacy, CPP/siRNA complex stability, and the properties of the CPPs, such as hemolytic activity, buffering capability and cell toxicity. As a result, we have introduced a new peptide with a completely redesigned N-terminus that displays adaptive response to its physical environment. This peptide – NickFect70 (NF70) – efficiently condenses siRNA, protects the cargo against degradation and effectively mediates target gene knockdown both in mammalian cell culture and in vivo, in a mouse model.
1. Introduction
Small interfering RNA (siRNA) molecules are able to regulate the expression of specific genes in cells through a process called RNA interference (RNAi).1 Use of an unmodified extracellular siRNA molecule is limited due to its native properties such as high density of negative charges, inadequate permeability through the cell membrane and high susceptibility to degradation in both intra- and extra-cellular environments.2 These RNA molecules have great therapeutic potential, although their use could significantly be enhanced by the application of a suitable delivery method.3Cell-penetrating peptides (CPPs) are relatively short peptides with the ability to gain access into the cell and mediate the delivery of cargo that is covalently or non-covalently attached to them.4 CPPs containing several positively charged amino acids (arginine, lysine, histidine and the non-proteogenic amino acid ornithine – O) in their sequence are adequately able to non-covalently associate with the negatively charged nucleic acid backbone. Complex formation occurs mainly via electrostatic interactions, and this strategy can be used to deliver a multitude of different molecules, including siRNAs.5,6 While this strategy is highly advantageous, the adequate stability and dissociation of the CPP/siRNA complexes need to be optimized.7,8 The CPP/cargo complexes mainly enter cells via endosomal pathways, and end up in the acidic endosomal compartments or in the lysosomes. Histidine-rich peptides are considered to be endosomolytic and have been exploited in the delivery of drugs, and also for different nanomedical applications.9
Designing delivery vectors with pH-dependent amphiphilicity and environmentally sensitive cargo release may offer an advantage in obtaining efficient delivery.10 Several amphipathic CPPs have been used to efficiently deliver siRNAs into mammalian cells,11 therefore we based our design on the amphipathic CPP NickFect55 (NF55). NF55 is a CPP and is a plasmid DNA delivery vector that has applications both in vitro and in vivo.12 It was initially designed for the delivery of plasmid DNA with optimal CPP/DNA complex stability and positive charges positioned along a theoretical alpha-helix. It has a relatively low +3 net charge, a fatty acid modification and a non-proteinogenic amino acid, ornithine, in the CPP sequence.12
Here, we have designed a series of CPPs with several distinct modifications, with the aim of increasing the charge, pH sensitivity, and delivery efficiency of NF55. Histidine is an amino acid that has a side chain pKa value of 6.0. Its side chain has a lower pKa than that of lysine (pKa 10.53) or arginine (pKa 12.48), and at a physiologic pH of 7.4 only a small fraction of the histidine molecules are protonated and donate to the hydrophobicity of the N-terminal. The difference in the side chain pKa can make peptides containing histidines tunable to the acidic conditions that are found intracellularly in endosomes and in lysosomes.13 The addition of a fatty acid tail and histidines to the peptide sequence creates two different regions in the CPP: a hydrophobic and a hydrophilic region. These regions support complex formation and stability, and they also prompt interactions with the cell membrane,8 as well as alternate the equilibrium between the cargo and the CPP in the complexes, depending on the environment.
We aimed to rationally design an efficient pH sensitive siRNA delivery vector that could transport cargo molecules into cells both in vitro and in vivo. For this, we modified (a) the peptide C-terminus with amino acid substitutions, (b) pH-sensitivity by the addition of histidines, (c) the lipophilicty of the peptide by the addition of histidines and a fatty acid, and (d) the distribution of histidines. We investigated how implementing these modifications affects the physico-chemical properties of the formed nanoparticles that are able to yield a high transfection efficacy.
2. Results and discussion
2.1. Design of pH-sensitive histidine containing NickFect peptides
In this study a series of NickFect (NF) peptides was designed for the delivery of siRNA (Table 1). To achieve this, the sequence of the CPP NF55 was modified (a) to make it pH sensitive, (b) to increase its positive net charge, (c) to maintain the theoretical alpha-helical structure in the C-terminal part of the peptide and to distribute the positive charges of the lysine molecules on one side of the theoretical alpha-helix, and (d) to optimize the hydrophobicity of the peptide.| CPP | Ca | FAb | Peptide sequence | Chargec | log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Dc Dc |
Gene knock-downd (%) | ||
|---|---|---|---|---|---|---|---|---|
| pH 7.4 | pH 6.0 | pH 7.4 | pH 6.0 | |||||
| a Peptide sequence. b The length of the saturated fatty acid added to the N-terminus of the peptide sequence (0 – w/o fatty acid; 10 – decanoyl; 18 – stearoyl; 20 – arachidyl; 22 – behaoyl). c Calculated with MarvinSketch, Chemaxon. d Maximum knock-down of luciferase gene, 25 nM siRNA, based on Fig. 1 and ESI Fig. 3.† e Synthesis continued from the sidechain amino group instead of α-amino group. f Lysine tree added to the Lys7 of the peptide. | ||||||||
| NF55 | — | 18 | AGYLLGOeINLKALAALAKAIL | 3.1 | 3.8 | 0.4 | −2 | 75 |
| PF6 | — | 18 | AGYLLGKfINLKALAALAKKIL | 8.9 | 10.9 | nd | nd | 84 |
| NF71 | H4 | 18 | HHYHHGOeILLKALKALAKAIL | 4.6 | 7.8 | −3.5 | −8 | 88 |
| NF712 | 20 | −2.5 | −7 | 83 | ||||
| NF713 | 22 | −1.5 | −6 | 47 | ||||
| NF700 | H6 | 0 | HHHHYHHGOeILLKALKALAKAIL | 5.2 | 9.3 | −12.5 | −17.5 | 6 |
| NF701 | 10 | −8.5 | −14 | 20 | ||||
| NF702 | 18 | −5.5 | −10 | 70 | ||||
| NF70 | 20 | −4.5 | −9 | 92 | ||||
| NF703 | 22 | −3.5 | −8 | 73 | ||||
| NF704 | H61 | 18 | HHHHHHGOeILLKALKALAKAIL | 5.3 | 9.4 | −7 | −12 | 82 |
| NF705 | 20 | −8 | −11 | 52 | ||||
| NF706 | 22 | −4 | −9 | 59 | ||||
| NF707 | H62 | 18 | HHHHHHYLLGOeILLKALKALAKAIL | 5.1 | 9.0 | −2 | −5 | 70 |
| NF708 | 20 | 0 | −4.5 | 71 | ||||
| NF709 | 22 | +1 | −3 | 55 | ||||
| NF721 | H8 | 18 | HHHHHHYHHGOeILLKALKALAKAIL | 5.9 | 10.8 | −7.5 | −11 | 55 |
| NF72 | 20 | −6.5 | −10 | 80 | ||||
| NF722 | 22 | −5.5 | −9 | 45 | ||||
Several studies have used histidine-containing peptides or liposomes modified with histidine-containing peptides previously, for the delivery of cargo into cells.14–16 Histidine residues have been included in the peptide sequence to increase their efficacy in the delivery of nucleic acids and to potentiate their endosomal escape.9,13 At physiologic pH the histidines are relatively uncharged and contribute to the hydrophobicity of the N-terminus. At low pH values the sidechain of the histidine becomes cationic upon protonation and increases the net positive charge of the peptide.17 Accordingly, at the pH found in the endosomal structures, the net charge of the histidine-containing peptides increases,13 making these delivery vectors responsive to acidic conditions as the interaction equilibrium between the CPP and the nucleic acid cargo changes.18,19 The location of the histidine-rich domain in the peptide sequence was shown to be critical for enhancing the transfection efficacy.20
In our design, to create defined hydrophobic and hydrophilic regions in the peptide NF55 C-terminus, one positive charge was added by replacing Ala14 with Lys along a theoretical α-helix, and the hydrophobicity was increased by the replacement of Asn9 with Leu. In the preliminary experiments we analyzed the impact of the histidine modification location, so histidines were added in either the NF55 C-terminal or the N-terminal part of the sequence. The Transportan10 (TP10) based histidine containing pH-sensitive peptides has been developed by other groups.21–23 When choosing the strategy for modifying the TP10 derived third generation peptide NF55, we tested both C-terminal and N-terminal modifications. In our case, the histidine modifications in the C-terminal part of NF55 led to a decrease in the delivery efficiency (ESI Fig. 1†). Therefore, 4, 6, and 8 histidine containing modifications in the N-terminus were included in the following design. The designed peptide contained three functional segments: an N-terminal fatty acid moiety, which is important for non-covalently formed nanoparticle formation and stabilization; an N-terminal hydrophobic/protonatable polyhistidine pH-sensitive switch; a C-terminal cationic peptide part with lysine molecules aligned on one side of a theoretical alpha-helix. Similar designs containing three functional parts in the same vector have been used in other works.10,16
In order to validate the position of the histidines and the importance of the tyrosine molecule, the H6-C20 peptide, a peptide with six histidines placed only on one side of the tyrosine (H62-C20), and one without Tyr (H61-C20), were included. Tyrosine can be considered to be an isosteric analogue to the uncharged form of histidine,24 and could stabilize the hydrophobicity of N-terminal peptide part.
Our experiments confirmed the importance and influence on transfection efficacy of Tyr in the H6 peptide (H6 vs. H61vs. H62) (ESI Fig. 3†). Interestingly, the H62 peptides were drastically less efficient and therefore excluded from the following experiments. This may be related to the significantly more positive logD values of the H62 analogs (Table 1).
We have shown earlier that a highly positive net charge and the presence of a fatty acid are important in complex formation between Transportan analogs and siRNA.19 Several groups have used a stearic acid modification in histidine containing peptides,16,25,26 but it has been shown that the length of the fatty acid chain plays a role in the transfection efficacy of nucleic acids by CPPs.27 Therefore, in order to optimize the hydrophobicity with the fatty acid tail, and to test the optimal conditions for each peptide sequence, saturated fatty acid modifications of C18, C20 or C22 were tested.
Additionally, in the H6 series, C10 and a peptide without a fatty acid (C0) modification were added. In the CPP PepFect14 the longest tested carbon chain of the fatty acid increased the bioavailability of splice correction oligonucleotides (SCO) in PF14/SCO complexes in the cells when compared with shorter chain modifications.27 In this work, however, an optimal fatty acid length had to be chosen for each corresponding histidine modified peptide (ESI Fig. 3†).
To model the pH sensitivity, lipophilicity and theoretical net charge of the peptide, pH vs. charge, and the calculated lipophilicity (logD) were recorded (Table 1). In addition, the buffering ability was tested by titration (ESI Fig. 2†). There was a correlation between the pH-dependent increase of the net charge and the buffering ability, depending on the number of the histidines in the sequence (ESI Fig. 2A and B†). However, we did not find a straightforward correlation between the logD values and the transfection efficacy, rather simply indications of the possible optimal range for each peptide sequence for nanoparticle formation (Table 1).
2.2. Gene knockdown by NF/siRNA complexes
In initial experiments all the new CPPs were compared for their efficacy to deliver siRNA into the U87-Luc cells and knockdown the reporter gene (ESI Fig. 3†). Accordingly, the best peptide was chosen for further experiments.In the down-regulation experiments, three CPPs, PF6, H4-C18 and H6-C20, were equally efficient at the CPP to siRNA molar ratio 30:1 (MR30). At MR40, H6-C20 slightly over performed the others. The CPP/siLuc complexes with PF6 and H4-C18 already achieved their maximum knockdown at MR20 (Fig. 1A). The 50% inhibition concentration (IC50) values at MR30 were lowest for the complexes formed with H4-C18, with an average of ∼7 nM siRNA concentration (Fig. 1B). The IC50 for the PF6/siRNA complexes were 1.5 times higher, with an average of ∼10 nM, and for the H6-C20 complexes the values were 2.2 times higher, with an average of ∼15 nM (Fig. 1B). The H4-C18 complexes had similar IC50 values of ∼11 nM to those of the PF6 complexes at the lower MR20 (ESI Fig. 4A†). In the case of the H6-C20 complexes, the increase in MR did not lead to a significant decrease in IC50 value (ESI Fig. 4B†). The H8-C20 complexes did not show any advantages over the H6-C20 or H4-C18 peptides (ESI Fig. 4C†). The difference in transfection efficacies of the H8-C20 and H6-C20 complexes may be related to the dissimilar self-assembly potential. The two histidines change the calculated logD value at pH 7.4 from −4.5 in H6-C20 to −6.5 in H8-C20 (Table 1).
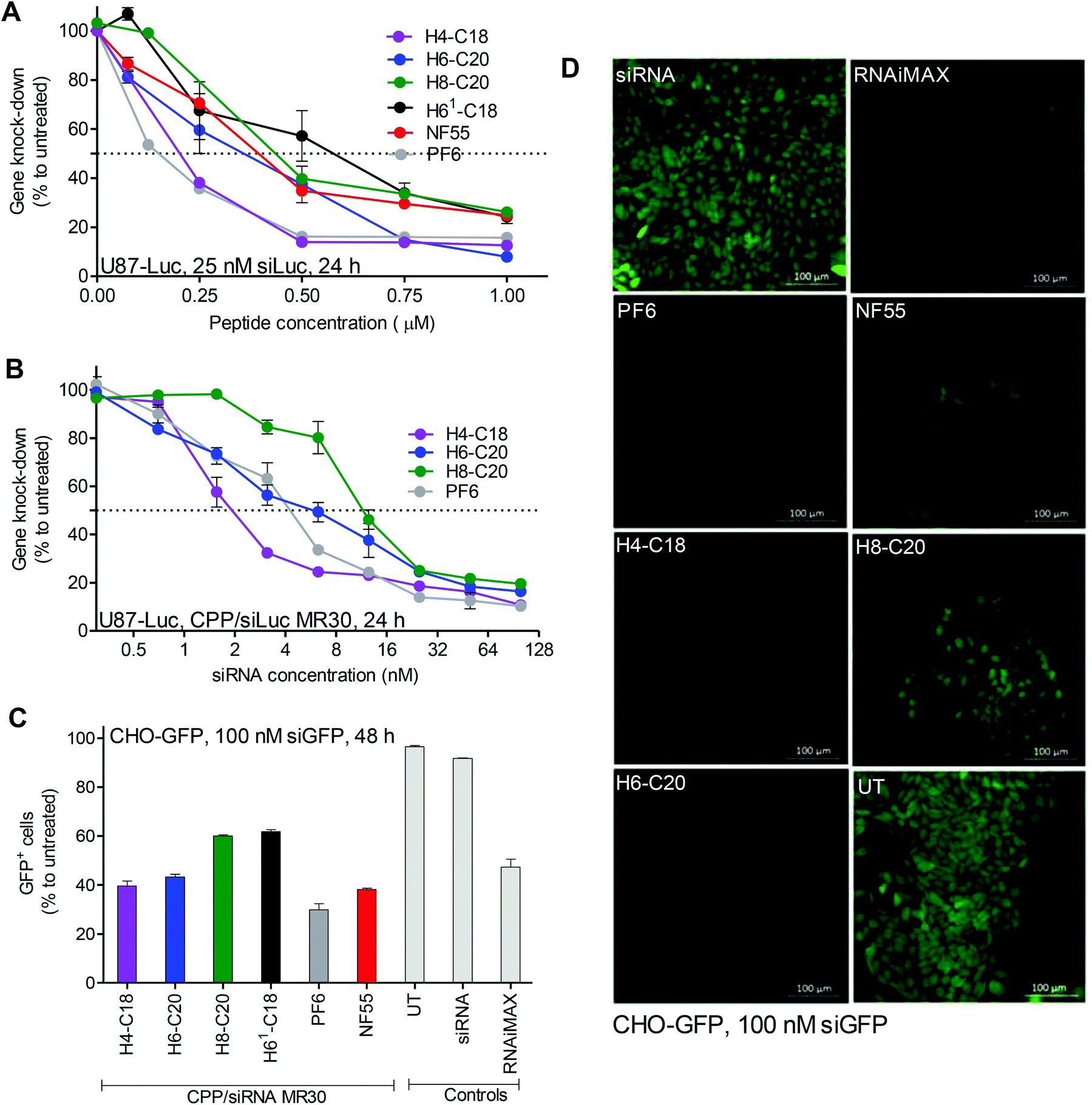 | ||
| Fig. 1 CPP/siRNA transfection efficacy and percentage of transfected cells. Experiments were done in serum (10% FBS) containing media. Luminescence from U87-Luc cells (A, B) form reaction with cell lysate 24 h post-transfection. (A) CPP to siRNA molar ratios (MRs) 1–40 were used with 25 nM siRNA final concentration in cell culture media. (B) CPP/siRNA complexes were formed at MR30 and siRNA final concentration was varied from 0.3 nM–100 nM. Fluorescence from CHO-GFP (C and D) cells was measured or detected 48 h post-treatment. Final concentration of siGFP on cells was 100 nM. (C) FACS analysis (GFP+ cell population) of transfected cells. FACS threshold was set at a very strict 0.1% of untreated cells, considered as GFP−, meaning that cells with downregulation but at least some remaining GFP were still considered GFP+. (D) Confocal microscopy images of live cells treated with CPP/siGFP complexes. Scale bars 100 μm. | ||
As indicated by the decrease in the size of the majority of nanoparticles from 284 nm to 175 nm, measured by DLS, when MR is increased from 30 to 40, the H8-C20 complexes may need a higher CPP to cargo ratio to form optimal self-assemblies that could lead to a higher transfection efficacy. This is also indicated by the increase of transfection efficacy in the H6-C20 and H8-C20 complexes when MR is increased (Fig. 1A). The H4-C18/siRNA nanoparticles may have reached their optimal self-assemblies already at lower MRs, as the transfection efficacy does not increase when the MR is higher. This shows that for each peptide, an optimal nanoparticle formation condition may be needed in order for it to achieve its maximal efficacy.16
The knockdown is probably pH-dependent, as indicated by the transfection in the buffers at pH 6.0 and 7.4 (ESI Fig. 4D†). Alternatively, it could be that the change of transfection efficiency is the result of changes in the nanoparticle size and shape, as well as due to interactions between the cell membrane and the nanoparticle.
In confocal microscopy images, the detected green fluorescent protein (GFP) signal was the lowest in cells treated with H4-C18/siRNA, H6-C20/siRNA and PF6/siRNA complexes (Fig. 1D). The FACS analysis revealed that the PF6/siRNA complexes were able to transfect and induce knockdown in approximately 70% of the cells. Complexes with NF55, H4-C18 and H6-C20 led to downregulation in 55–60% of the cells. H61-C18 and H8-C20 had the lowest GFP− cell population, with 40% (Fig. 1C) of the cells with the GFP signal below the given threshold.
An MTS toxicity assay on cells showed that most of the used CPP/siRNA complexes were not toxic to cells (ESI Fig. 5B†). The H4-C18 complexes were only more toxic from concentrations above 5 μM, and the H61-C18 complexes were toxic at 10 μM concentrations (ESI Fig. 5A†).
According to the obtained results, from the series two peptides stood-out. H6-C20 (NF70) was considered to be the most efficient, as it led to an efficient down-regulation of the reporter gene and did not show any significant toxic effects to the cells even at high concentrations. The second best was H4-C18, which had a significantly lower IC50 value, but was also toxic at higher peptide concentrations.
2.3. Characterization of CPP and CPP/siRNA nanoparticles
The positive charges in the used sequence of the CPPs drives complex formation with negatively charged nucleic acids, including siRNAs, and hydrophobic regions contribute to the interaction and formation of nanocomplexes with a specific size, shape and stability. The ability to form stable complexes depends on the peptide sequence, its net charge, its hydrophilicity/hydrophobicity profile, as well as the type of nucleic acid cargo molecule (DNA vs. RNA) and its molecular weight.28,29 It has been shown that the size, aspect ratio and dimensions of the particle influence both the cellular uptake and intracellular fate of the particles.30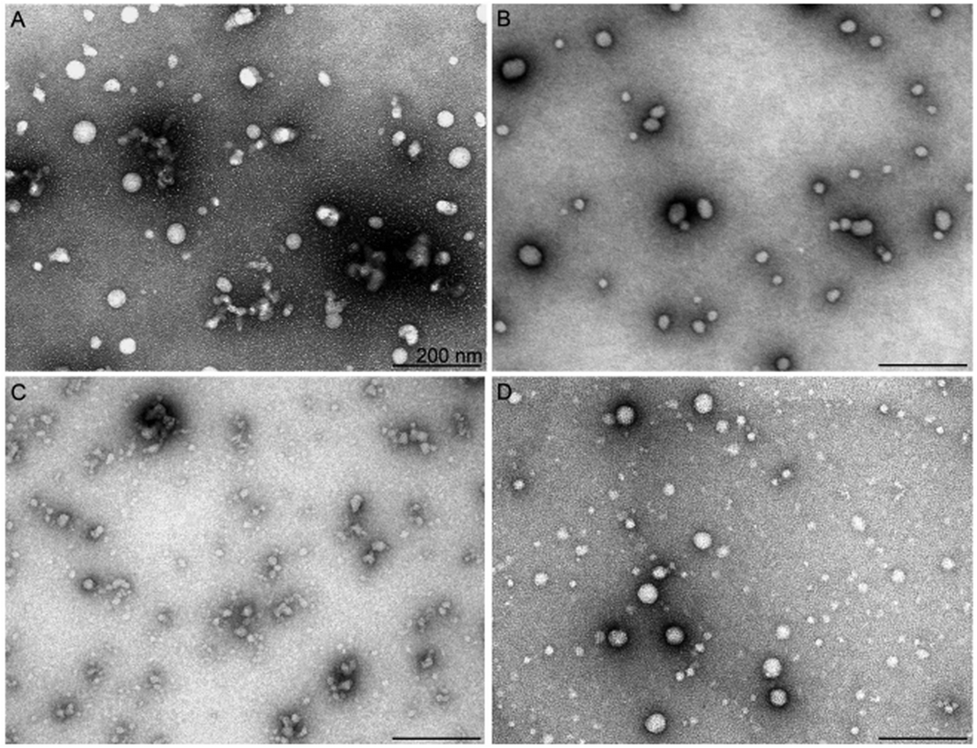 | ||
| Fig. 2 Shape and size of CPP/siRNA nanoparticles assessed by TEM. Negative staining tem images of CPP/siRNA nanoparticles. The nanoparticles were formed at MR30 in water and stained with aqueous uranyl acetate solution, and imaged at 13000× magnification. Scale bars: 200 nm. Nanoparticles formed with (a) H6-C20, (b) H4-C18, (c) H8-C20, and (d) PF6. | ||
In the absence of the fatty acid modification, H6-C0, the NPs did not reveal any significant luciferase silencing activity (ESI Fig. 3†). Upon increasing the length of the fatty acid on the peptide N-terminus, the acquired siRNA effect increased up to 95% of gene knockdown in the case of H6-C20. After exceeding the optimal fatty acid length, the RNA interference effect decreased, as observed with the H6-C22/siRNA nanocomplexes. TEM analysis corroborated a good correlation of activity with the morphology of the respective NPs. The H6-C0 peptide formed very few stable particles and detectable nanoparticles with siRNA. The observed particles were elongated, at >500 nm in length, and seemed to be loosely packed, as suggested by the high diameter of the complexes (ESI Fig. 6A†). The number of formed stable NPs was substantially higher in the H6-C10 and H6-C18 complexes. Furthermore, these were remarkably shorter/smaller (200–400 nm) and had a lower diameter (15–20 nm), suggesting a tighter packing and higher stability of the nanoparticles (ESI Fig. 6B and C†) that is in very good accordance with their biological effect (ESI Fig. 1†). H6-C22 packed siRNA mostly into very small particles that may have associated to form larger conglomerates of 10–20 nm (ESI Fig. 6D†). Analogous small nanoparticles and their conglomerates also formed upon condensation of siRNA with H8-C20 (Fig. 2C). Although the fatty acid in this peptide has the same length as in the most efficient peptide of this series, NF70, the formed particles were remarkably different, implying that two additional histidine residues in the former might add more hydrophobicity/charge to the N-terminal of the peptide than is required for optimal siRNA delivery. In the DLS of the H6 series with the fatty acid C10 to C22, nanoparticles with average sizes between ∼100–300 nm were formed (ESI Table 1†).
The second best CPP, H4-C18 (NF71), condensed siRNA to rather similar particles to H6-C20; however, their complexes were slightly less regular/spherical and homogeneous, ranging from about 20 to 60 nm in diameter in the TEM (Fig. 2) and with an average size of ∼180 nm in the DLS (ESI Table 1†). This implies a lower condensing capacity the shorter the chain of the fatty acid (C18 vs. C20) and that less histidine moieties are present in this peptide compared to H6-C20.
The predecessor of the histidine-rich series of peptides, NF55,12 which is a very efficient cellular delivery vehicle for pDNA, mostly formed spherical NPs at varying sizes of 10–50 nm and also elongated conglomerates of up to 100 nm in length (ESI Fig. 6E†). The association of NP morphology with their gene silencing effect suggests that more homogeneous particles of about 30–50 nm in size enabled the most efficient downregulation of the target gene, and either too small or too large NPs, or highly heterogeneous ones, lead to a moderate or poor siRNA effect in cell culture conditions.
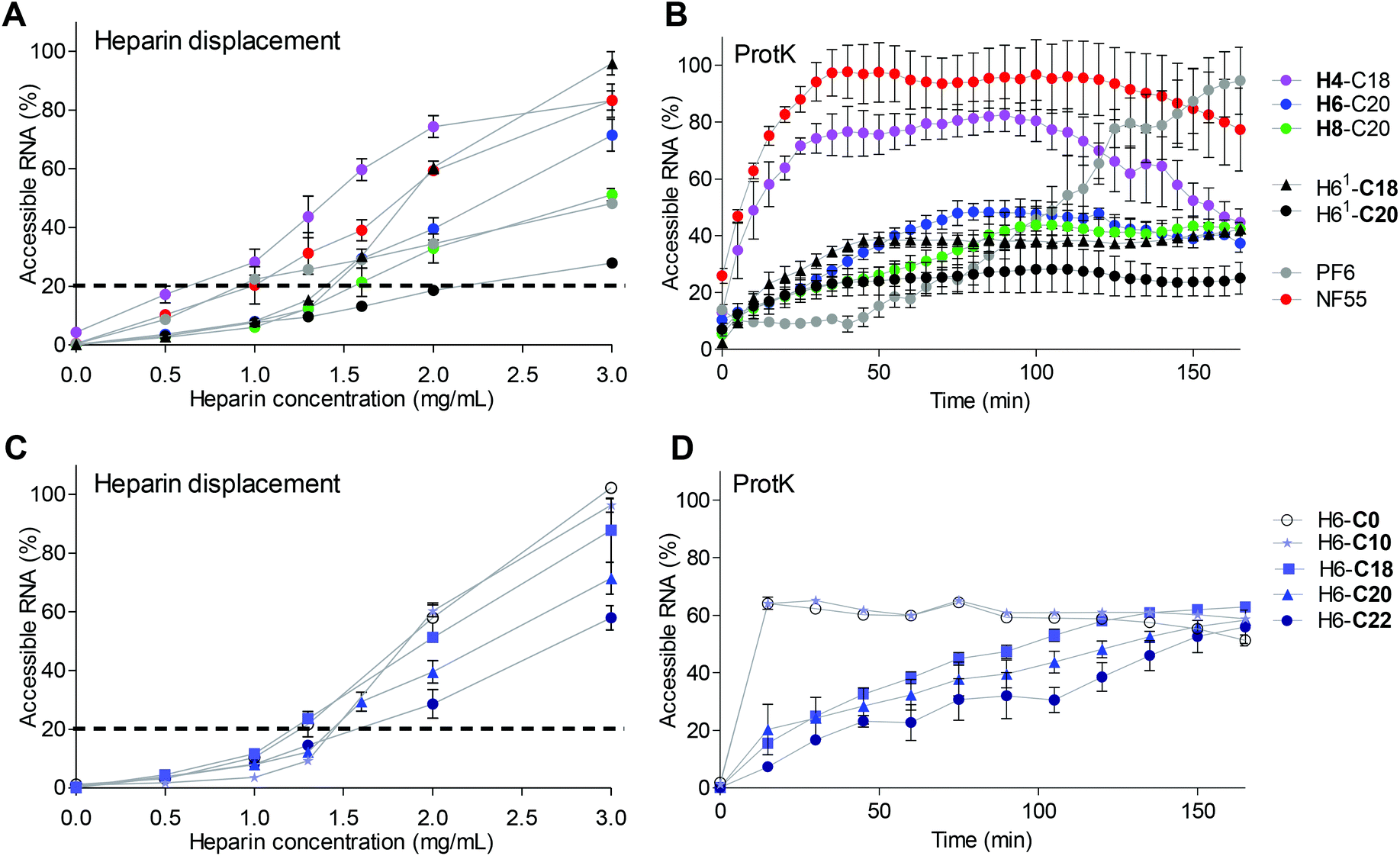 | ||
| Fig. 3 Analysis of CPP/siRNA complex stability and packing. (A) Heparin displacement assay. CPP/siRNA complexes formed at MR30. Heparin solution co-incubated with the complexes at 37 °C for 30 min. Results are shown as % to free siRNA at the same concentration as used in the complex solution. (B) Stability of complexes to proteinase K degradation (>14.4 U per well) measured as binding of fluorescent dye PG to nucleic acid over a time-period of 180 min. Results are normalized to free siRNA. (C) Heparin displacement of H6 series peptides with different fatty acid length. (D) Stability of H6 series to proteinase K. | ||
Interestingly, H4-C18 had 17% of siRNA in the complexes accessible already at 0.5 mg ml−1 heparin concentration compared to 10% in the NF55/siRNA complex samples (Fig. 3A). The stability of the complexes containing H4-C18, NF70 and H8-C20 showed an increase in stability depending on the number of histidines included in the peptide sequence (Fig. 3A). Furthermore, the complexes formed with H61-C20 lacking the tyrosine in the N-terminal peptide part (Table 1) had significantly higher stability to heparin displacement than the H6-C20 peptide and even the H8-C20 peptide. The stability may be caused by the higher affinity of uninterrupted histidine span to siRNA compared to the tyrosine containing motif. It has also been shown earlier that the uninterrupted histidine sequence in the stearyl-TP10 sequence significantly increases the affinity between the peptide and the splice correcting oligonucleotide.26
When comparing the H6 peptide series with different fatty acid modifications, there was a correlation between the stability of the complex and the fatty acid length. Longer fatty acids packed the siRNA into NPs with a higher stability in the heparin displacement assay (Fig. 3C) and against enzymatic degradation (Fig. 3D). The peptide analogue with C22 was the most resistant to heparin, and even 3 mg ml−1 heparin concentration resulted in only 60% siRNA displacement (Fig. 3C). Still, as indicated by lower transfection efficacy, an optimal hydrophobic/hydrophilic ratio might have been exceeded.
The complexes with PF6 had a significantly lower stability to longer exposure to the enzyme (Fig. 3B), than complexes containing H6-C20 and H8-C20 peptides. The H4-C18/siRNA complexes were only slightly more resistant than the NF55/siRNA complexes to proteinase K. Intriguingly, the complexes with H61-C18 that had higher transfection efficacy had a similar stability to complexes formed with H6-C20. The H61-C18 analogue was less stable, but also achieved a higher transfection efficacy than H61-C20. This indicates once again that the histidine sequence vs. fatty acid length has to be optimized accordingly. The uninterrupted histidine sequence in the H61 analogue may stabilize or strengthen the interactions with the cargo (H61-C20 vs. H6-C20), whereas the shorter fatty acid may result in more dynamic particles (H61-C18 vs. H61-C20) (Fig. 3B).
When comparing stability and transfection efficacy, there is an optimal range of stability that the nanoparticle has to fall into. The CPP has to be able to pack and protect the siRNA from the degradation, however it also has to be able to release the cargo at the target site. Interestingly, both H4-C18 and NF70 have similar theoretical logD values (Table 1), but as indicated by the lower efficacy of H6-C22, there might be a slight variation in the range for each sequence.
NPs with the H8-C20 peptide may be too stable in order for the siRNA to be released, as indicated by the lower transfection efficacy (Fig. 1 and 3). NF70/siRNA nanoparticles have a stability profile between that of the H4-C18/siRNA and H8-C20/siRNA nanoparticles, and the highest gene knockdown (Fig. 1 and 3). The increase in the number of histidines increases the pH-buffering ability of the peptide, although for delivery purposes there might be a cutoff for the histidine chain length, as H8 peptides were not as efficient as H6 or H4 peptides (Fig. 1). For the tested histidine-rich peptides, there was a pH-dependent efficiency in knockdown (ESI Fig. 4D†), as for the complexes formed with PF6, a known pH-dependent delivery CPP.
The H4-C18/siLuc complexes led to efficient knockdown even at MR20 and had a low IC50, but the complexes formed with this peptide were significantly less stable than those with H6-C20 (Fig. 1A, 3A, and B). NF70 probably has an optimal stability profile, as the peptides with higher stability show lower transfection efficacy (Fig. 1 and 3B).
Red blood cells have been used for assessing ex vivo the membrane activity and also the endosomolytic properties of pH-responsive delivery agents.36 Interestingly in H6-C20 complexes, the hemolytic IC50 concentration was 2.5 times higher than that of NF55. A comparison of the H6-C20 and H61-C20 complexes that both have six histidines showed a significantly lower IC50 for H61-C20, probably due to the difference of histidine distribution in the sequence. H8-C20 was drastically less hemolytic than other peptides and did not show any significant membrane activity even at 10 μM peptide concentration (Fig. 4A). The addition of siRNA to the peptide solution and formation of the respective complexes decreased the hemolytic activity approximately two fold (ESI Table 2†), suggesting that the membrane activity could be mediated by the free or accessible CPP that has not been neutralized by the interaction with the negatively charged siRNA.
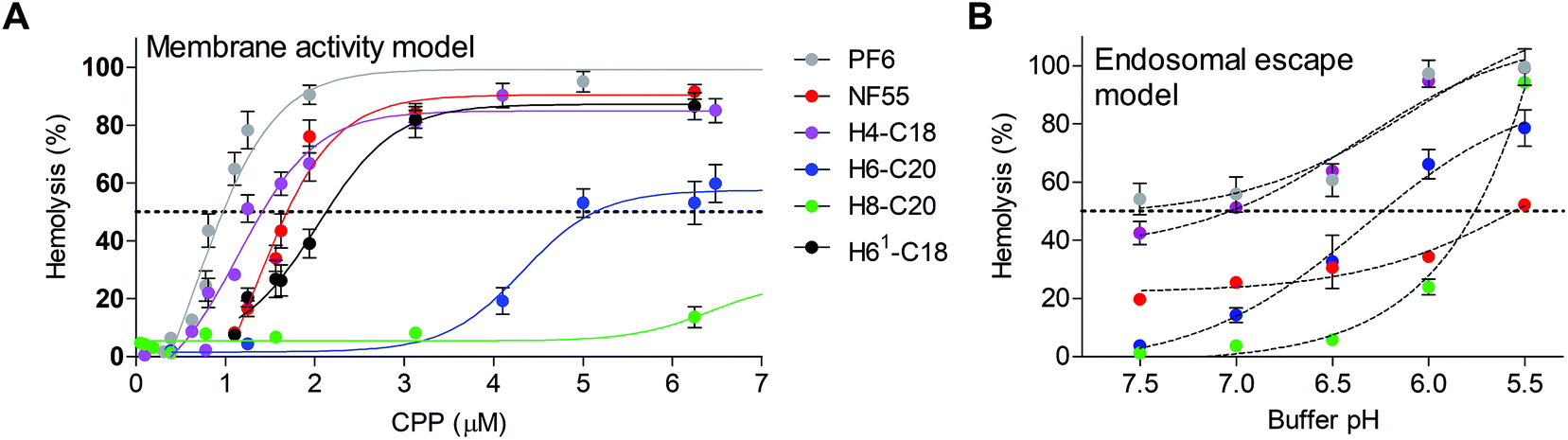 | ||
| Fig. 4 Membrane activity of nickfect peptides. Hemolytic activity of peptides was measured after 1 h-incubation of murine red blood cells with peptide solutions at 37 °C. After incubation released hemoglobin was quantified in the supernatant by measuring absorbance at 540 nm. (a) Cells in PBS pH 7.5 supplemented with 5% glucose at 37 °C. as controls RBC in 2× PBS treated with 0.1% Triton X-100 with 5% glucose (100% hemolysis) and mQ with 5% glucose (0% hemolysis) were used. (b) Modeling of endosomolytic activity of CPPs using buffer solutions with different pH. Peptides were used at 0.75 μm concentrations. Hemolytic activity was assessed in PBS buffer at pH 5.5–7.5. | ||
A modified version of the protocol was used to analyze the endosomolytic activity of the NFs at a peptide concentration of 0.75 μM (corresponding to the CPP concentration in complexes formed at MR20). As expected, NF55 was the least influenced by the pH decrease and the hemolytic activity only increased at pH 5.5 by 25% (Fig. 4B). The highest pH dependent increase of hemolytic activity was exhibited by H8-C20. It had almost no effect at physiologic pH, but lysed almost all cells at pH 5.5, and its hemolytic activity started to increase at pH 6.0. A similar tendency was also detected for H6-C20, but the increase was less drastic. It should be noted that the damage of RBC membranes was significantly higher in a lower pH buffer even in the case of a negative control, i.e. peptide-free solution (1× PBS and glucose, pH 5.5) (Fig. 4B).
Out of the tested peptides, NF70 had an advantageous pH dependent membrane activity profile. It had a low membrane activity at physiologic pH and increased in activity at low pH (Fig. 4) accompanied with an efficient gene knockdown (Fig. 1). In H4-C18 complexes, the high transfection efficiency may be caused mainly by the membrane activity of the peptide (Fig. 4) or the negative effect on the cell proliferation at higher concentrations (ESI Fig. 5†), as the formed complexes are not as stable as the H6-C20/siRNA particles (Fig. 3A and B).
The authors note that although the hemolytic assay has been used as a viability assay, it is used in our context as a membrane activity model only. The hemolytic activity is totally abolished in cell media and especially if serum is added as a media supplement (data not shown). Furthermore, the hemolytic activity does not correlate with the toxicity measurement results (Fig. 4 and ESI Fig. 5†).
2.4 In vivo gene knockdown with NF70/siFVII
As a target blood coagulation factor, VII mRNA was chosen. This vitamin K dependent enzyme belongs to the blood coagulation cascade and is mainly synthesized in the liver cells. It has a relatively short half-life compared to other clotting factors. H6-C20 was able to deliver siFVII to the mouse liver in vivo and a single injection resulted in an almost 60% knockdown (Fig. 5). Compared to siRNA delivery with nanogel nanoparticles found in other work by another group,37 30% knockdown was achieved, compared to the siRNA treated group, with a significantly lower siRNA dose (1.6 mg kg−1vs. 4 mg kg−1). Compared to the free siRNA, and commercially available in vivo transfection reagent In vivo JetPEI (Polyplus, Germany) at N/P 6, the NF70/siFVII complexes were able to mediate higher knockdown at the same dose.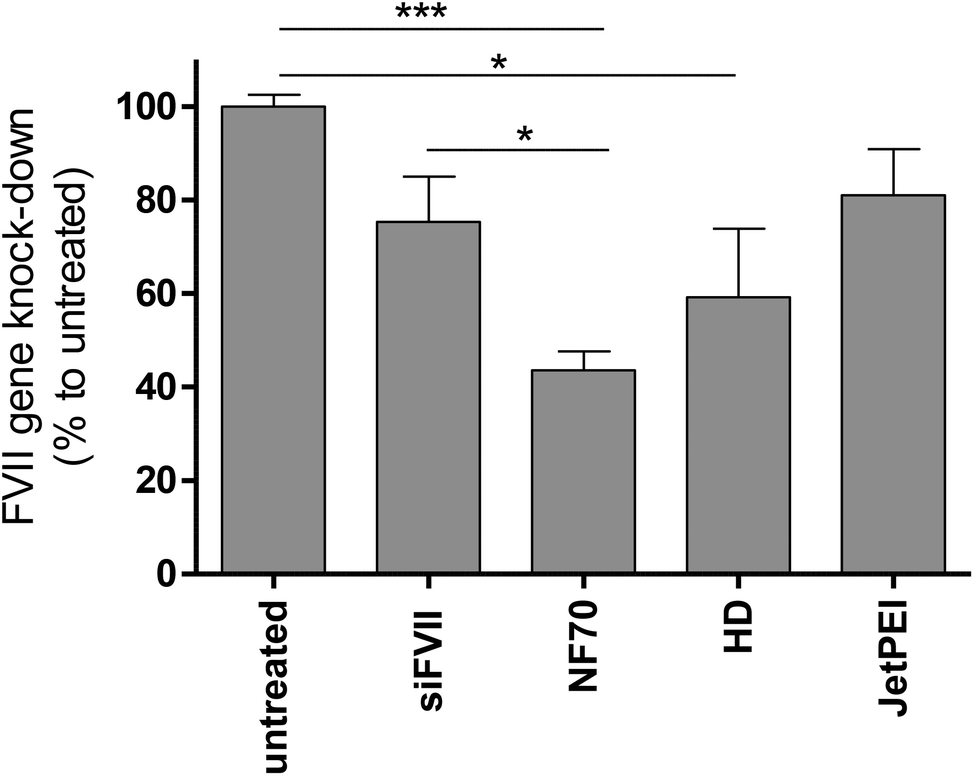 | ||
| Fig. 5 Supression of blood coagulation factor VII expression by H6-C20/siFVII complexes in vivo. The downregulation was assessed by qRT-PCR from liver tissues of Balb/c mice 48 h post-injection of NF70/siRNA (H6-C20) complexes with siRNA 1.6 mg kg−1 dose and MR30. NF70/siRNA, siRNA and UT n ≥ 4, JetPEI/siRNA and hydrodynamic (HD) n = 2. Statistical analysis one way ANOVA, p < 0.05, control groups untreated or siRNA. | ||
3. Conclusions
Delivery vectors that are pH-sensitive may enhance nanoparticle accumulation to lower pH regions in vivo, and the release of nanoparticles or siRNA from entrapment in endosomal compartments. In this work we have demonstrated an appealing, simple and efficient strategy to enhance and optimize the delivery efficacy and pH-sensitivity of a CPP-based delivery vector. We designed and tested pH-sensitive histidine-rich analogs of CPP NF55 with the ability to non-covalently associate a cargo of siRNA to form stable nanoparticles that are able to efficiently transfect cells.We showed the effect and importance of amino acid histidine number, their position and distribution in the peptide sequence and also the significance of a fatty acid modification and its length to the assembly and characteristics of the formed CPP/siRNA nanoparticles. We accomplished the design of a highly potent delivery vector for nucleic acid delivery, CPP NF70. NF70 is a pH-activated, histidine-rich CPP that has low cell toxicity, low membrane activity at physiologic pH, and forms stable nanoparticles. It successfully achieves gene knockdown both in vitro and in vivo.
4 Materials and methods
4.1 Peptide modelling
All peptide sequences were modelled and the charge or logD calculations were performed using MarvinSketch 15.9.14, ChemAxon Ltd, USA.
4.2 Cell culture maintenance
U87 MG-luc2 human glioma cells, stably expressing luciferase and U87 cells were grown on gelatinized (0.1% gelatine, Naxo, Estonia) dishes at 37 °C and 5% CO2 in Dulbecco's Modified Eagle's Medium (DMEM) (Sigma, Germany), and CHO stably expressing GFP were grown at 37 °C with 5% CO2 in Ham's F12 nutrient mixture (Capricorn, Thermo Fisher Scientific, USA). Cell media were supplemented with 0.1 mM non-essential amino acids, 1.0 mM sodium pyruvate, 10% fetal bovine serum (FBS) (Sigma, Germany), 100 U ml−1 penicillin and 100 mg ml−1 streptomycin (Invitrogen, Sweden).4.3 Solid phase peptide synthesis
Peptides were synthesized on an automated peptide synthesizer (Biotage Initiator+ Alstra) using the fluorenylmethyloxycarbonyl (Fmoc) solid phase peptide synthesis strategy with Rink-amide ChemMatrix resin (0.45 mmol g−1 loading) to obtain C-terminally amidated peptides. The fatty acid was coupled manually to the N-terminus of the peptide overnight, at room temperature with 5 eq. fatty acid. For the synthesis of the ornithine containing peptides, Boc-L-Orn(Fmoc)-OH (Iris Biotech, Germany) was used. The reaction was carried out using HOBT/HBTU as coupling reagents in DMF, with DIEA as an activator base. For the synthesis of PF6, chloroquine analogs (trifluoromethyl quinoline derivative) were coupled to the lysine tree over succinic anhydride (Sigma-Aldrich) added to the Lys7 (with Mtt sidechain protecting group removed with 0.1%TFA and 3%TIS in DCM. Fmoc-Lys (Fmoc)(Iris Biotech)) of the peptide.Cleavage was performed with trifluoroacetic acid, 2.5% triisopropylsilane and 2.5% water for 2 h at room temperature. Peptides were purified by reversed-phase high-performance liquid chromatography on a C4 column (Phenomenex Jupiter C4, 5 μm, 300 Å, 250 × 10 mm) using a gradient of acetonitrile/water containing 0.1% TFA. The molecular weight of the peptides was analyzed by matrix-assisted laser desorption-ionization/time of flight mass spectrometry (Brucker Microflex LT/SH, USA). The concentration of the peptides was determined based on dilutions of accurately weighed substances and absorption of tyrosine, where applicable.
4.4 Peptide environment pH maintenance assay by titration
To assess the ability of the peptides to maintain the pH of the solution in a certain range (due to the histidines being added to the sequence), 5 ml of a 200 μM peptide solution in mQ water was prepared and 1 M TFA was added to achieve the same pH 2.5 for all peptide solutions. 0.5 M NaOH (5 μl) solution was added while stirring and the pH was measured after each step. The change in pH was documented and the titration curve is shown as a graph (pH vs. added NaOH solution).4.5 The formation of peptide/siRNA complexes and experiments in cells: downregulation of the reporter gene, fluorescence-assisted cell sorting (FACS), and confocal microscopy
For assessing the siRNA mediated RNAi, CHO-GFP or U87-luc2 cells (5 × 105 cells per well) were seeded on a 24-well plate a day prior to the experiment and grown in serum containing media. On the experiment day, prior to adding pre-formed complexes (1/10th of the final volume per well), the media was replaced with fresh serum containing medium.For complex formation, the peptide was mixed with luc2 siRNA (sense 5′-GGACGAGGACGAGCACUUCTT-3′, antisense 5′-GAAGUGCUCGUCCUCGUCCUU 3′, Metabion, Germany) or with siRNA against GFP (5′-GGCUACGUCCAGGAGCGCACC, 3′-UGCGCUCCUGGACGUAGCCUU) in mQ water (pH 5.3–6.3) using the peptide to siRNA molar ratios (MR CPP:siRNA) up to 40:1 and incubated at RT for 1 h. For siLuc2, the final concentration in the media was 25 nM and for siGFP it was 100 nM. The cells were co-incubated with complexes for 4 h (37 °C and 5% CO2), and thereafter fresh serum containing media was added and followed by further incubation (20 h for siLuc2 or 44 h for siGFP complexes). Luciferase gene downregulation was measured from cell lysates with the Promega luciferase assay system (Promega, Sweden) in combination with a GLOMAX 96 microplate luminometer equipped with GLOMAX 1.9.2 software (Promega). The results were normalized to the protein content of each sample (DC Protein Assay, BioRad, USA).
For the determination of IC50, complexes were formed at MR30 and diluted before addition on cells to the final siRNA concentrations of 100, 50, 25, 12.5, 6.25, 3.13, 1.6, 0.7 and 0.3 nM in wells. Luciferase activity was determined as described in the previous paragraph.
For analyzing the effect of pH on the transfection efficiency the complexes were formed at MR30, incubated and diluted before addition to the U87-luc cells to obtain an siRNA final concentration of 10 nM in transfection media. HEPES buffer supplemented with 10% FBS was used as the transfection media. The complexes were co-incubated with cells for 4 h and then the media was replaced with cell culture media. After a further 20 h, the incubated cells were lysed and analyzed as described previously.
For flow cytometry analysis (FACS), the CHO-GFP cells were seeded and treated as described previously. After incubation, the cells were washed with 1× PBS and detached from the plates with trypsin-EDTA (0.25%), and re-suspended in PBS supplemented with FBS (5%) and transferred to transparent U-bottom 96-well plates. FACS was carried out with a BD LSR II flow cytometer (BD Biosciences, San Jose, CA, USA) equipped with a 488 nm argon laser. The population of viable cells was determined from a scatter plot: forward scattered plot (FSC) vs. side scattered light (SSC) plot. A minimum of 10000 events from the viable cell population per sample were analyzed using the BD FACS Diva software. Results are shown as the population with GFP signal lower than that of 0.1% of untreated cells.
For confocal microscopy, 2 × 105 cells per chamber were seeded on chambered coverglass (NUNC 8-chamber plates). After treatment with the complexes (25 μl of pre-formed CPP/siRNA complexes added to cells in 225 μl serum containing media) and incubation (48 h), GFP fluorescence images of the cells were captured using the confocal microscope Zeiss LSM 710 with the excitation at 488 nm (493–616 filter).
4.6 Negative staining and visualisation of complexes by TEM
For the morphological analysis the CPP/siRNA complexes were assembled as described above at MR30. For negative staining, copper grids were covered with a Formvar film and a carbon layer using a Leica EM ACE600 carbon coater (Leica Microsystems, Germany). Then, 10 μL of each sample was absorbed onto the grids for 2 min and any excess of the solution was gently removed. The samples were then exposed to 2% aqueous uranyl acetate solution for 1 min. After removing the excess stain with filter paper, the samples were allowed to air-dry. The samples were examined at 120 kV accelerating voltage on an FEI Tecnai G2 Spirit electron microscope (FEI, The Netherlands).4.7 Dynamic light scattering (DLS) measurements
The hydrodynamic mean diameter of the nanocomplexes was determined by dynamic light scattering studies using a Zetasizer Nano ZS apparatus (Malvern Instruments, UK). The peptide/siRNA complexes were prepared in milli-q water, as described for transfection. All results were based on three measurements from four independent samples. All data was converted to “relative by intensity” plots from where the mean hydrodynamic diameter was derived. The complex solutions were formed in 1/10th of final volume and diluted before measurements.4.8 Membrane activity assay on murine red blood cells: hemolysis
For evaluation of the membrane activity of the peptides, a hemolysis assay was used. For this, whole blood was collected from a mouse (adult, BALB/c) saphenous vein into a heparinized collection tube. The blood was centrifuged (2300g, 15 min) to separate the cells (red blood cells – RBC) from the plasma. The RBCs were washed according to the following procedure.The RBCs were washed 3× with 10 ml 0.9% NaCl saline. After each step, the cells were centrifuged (600g, 15 min) and the supernatant was discarded. The RBCs were re-suspended in 5 ml saline and centrifuged (600g, 15 min). The cells were washed and re-suspended in 2× PBS with the needed pH (pH 5.5–7.5). To each 100 μl of peptide solution, 100 μl of 10% glucose was added, mixed, and an equal volume (200 μl) of RBC in 2× PBS was added and gently mixed. The peptide solution with RBC-s was incubated at 37 °C for 1 h with gentle mixing (max 300 rpm) and after that, the samples were centrifuged to pellet intact cells from the solution. Hemoglobin from the lysed cells was determined from each sample from the supernatant on transparent 96-well plates by measuring the absorbance at 540 nm.
Controls were incubated in a similar manner to the samples. As a non-hemolysis control, mQ supplemented with glucose (0%) was used and for the hemolytic control and 0.1% TritonX-100 with glucose (100%) was used (cells are suspended in 2× PBS before addition). Final percent was calculated as follows:
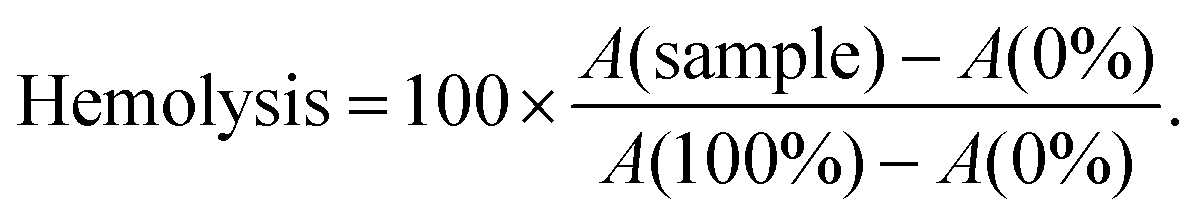
For hemolytic concentration determination, 2× PBS at pH 7.5 was used for incubation and 1× PBS at pH 7.5 was used for washing. Everything else is as described in previous paragraph.
4.9 MTS toxicity assay
Cell proliferation was analyzed with the CellTiter 96® Aqueous Non-Radioactive Cell Proliferation Assay (MTS) (Promega Biotech AB, Sweden) according to the manufacturer's instructions. For this 1 × 105 U87 cells were seeded 1 day prior to experiment on transparent 96-well plates. On experiment day, media was replaced with 90 μl of fresh media and to the cell complexes formed at MR30 (100 nM final siRNA concentrations) or free peptides at the range of 0.5–4 μM, final concentrations were added. Cells were treated with complexes for 20 h. The absorbance of formazan product was measured at 490 nm with Tecan Sunrise microplate absorbance reader (Tecan Group Ltd, Switzerland) and the percentage of viable cells was calculated using the GraphPad Prism software 5.0 (Graphpad Software, CA, USA).4.10 Fluorescent dye intercalation assay
Complexation of siRNA and CPPs was assessed by the Quant-iT™ PicoGreen® (PG) (Thermo Fisher Scientific, USA) assay in mQ water. Complexes were prepared as described previously, following the addition of diluted PG. For detection, the complexes (1/5 of volume), water (3/5) and PG (1/5) working dilution were incubated for 5 minutes and fluorescence was measured by a fluorimeter (λex = 492 nm, λem = 535 nm) (SynergyMx, BioTek). For the heparin displacement, instead of water, heparin salt solutions at different concentrations were used and the complexes were incubated at 37 °C for 30 min before addition of the PG dilution. Complex formation was also confirmed by gel electrophoresis.To assess the protection of siRNA from enzymatic degradation, after the addition of PG dilution and the initial measurement to set a starting point, Proteinase K (Thermo Scientific, USA) was added (>14 U ml−1, 20 μl of mixture per well containing dilution of 120 μl of enzyme in 1 ml), co-incubated with the complexes and the fluorescence was measured over a 10 h period.
4.11 In vivo experiments
For the gene downregulation studies, male Balb/c, 2–4 month old mice were used, with 2–5 mice per group. The CPP/siRNA complexes were formed at MR30 and for each mouse a 1.6 mg kg−1 siRNA dose was used. Particle formulations were administered intravenously via tail vein injection. 48 h post treatment, liver tissues were harvested and snap-frozen for analyses. The tissues were homogenized using the Precellys®24-Dual homogenization system (Bertin Technologies, France) with the TRIzol reagent (Thermo Fisher Scientific). RNA was extracted according to TRIzol manufacturer's protocol with slight modifications. From the RNA, complementary DNA was synthesized using SuperScript IV Reverse Transcriptase (Thermo Fisher Scientific) according to the manufacturer's protocol. The synthesized cDNA was used for qPCR with 5× HOT FIREPol EvaGreen qPCR Supermix (Solis Biodyne, Estonia), according to manufacturer's protocol. The obtained data was analyzed using the 2−ΔΔCT method and normalized to untreated mouse group (100%). siRNA against blood coagulation factor VII (siFVII) sense 5′-gga(2′F -U)(2′F -C)a(2′F -U)(2′F -C)(2′F -U)(2′F -C)aag(2′F -U)(2′F -C)(2′F -U)(2′F -U)a(2′F -C)T*T-3′ and antisense 5′-g(2′F -U)aaga(2′F -C)(2′F -U)(2′F -U)gaga(2′F -U)ga(2′F -U)(2′F -C)(2′F -C)T *T 3′. The primers for PCR (Mus blood coagulation factor VII) forward ACAAGTCTTACGTCTGCTTCT, reverse: CACAGATCAGCTGCTCATTCT and (Mus Actin beta) forward: CCACACCCGCCACCAGTTCG and reverse: TACAGCCCGGGGAGCATCGT. The siRNA and primers were synthesized by the same manufacturer (Metabion, Germany). Data was collected with the ViiA 7 RT_PCR system and analysis was based on 2−ΔΔCT and normalized to the results from the untreated mice.For NF70/siRNA and the In vivo JetPEI/siRNA complexes, the same solutions were used for mixing and glucose final concentration 5%.
As a control, a hydrodynamic injection of 1 ml of siRNA was used and untreated mice were included. All animal experiments and procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of University of Tartu and were approved by the Estonian Laboratory Animal Ethics Committee (approvals no 81, dated Apr 04, 2016, and 69 and 70, dated Feb 9, 2011).
4.12 Statistical analysis
Statistical analyses of cell culture, fluorescent label intercalation assay and in vivo experiments were done on the GraphPad Prism software 5.0 (Graphpad Software, CA, USA). All results are shown as a mean with the SEM of at least three separate experiments, if not indicated otherwise.Data availability statement
The data used to support the findings of this study are available from the corresponding author upon request.Abbreviations
| CPP | Cell-penetrating peptide |
| PG | PicoGreen |
| NF | NickFect |
| PF | PepFect |
| MR | Molar ratio |
| FACS | Fluorescence assisted cell sorter |
| TEM | Transmission electron microscopy |
| siRNA | Small interfering RNA |
| O | Amino acid ornithine |
| Cn | Fatty acid carbon chain length |
| NP | Nanoparticle |
| TP10 | Transportan10 |
| RBC | Mouse red blood cells |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the European Regional Development Fund through the projects Tumor-Tech (3.2.1001.11-0008) and Center of Excellence in Molecular Cell Engineering (2014-2020.4.01.15-0013) by the Estonian Ministry of Education and Research (0180019s11) and Estonian Research Council (IUT20-26 and PUT1617). U87 cells were obtained from Caliper LifeSciences, USA; Fetal Bovine Serum: Gibco™ 1027010, Thermo Fisher Scientific, Sweden.References
- P. D. Zamore, Nature, 2007, 446, 864–865 CrossRef CAS PubMed.
- Y. Ren, S. Hauert, J. H. Lo and S. N. Bhatia, ACS Nano, 2012, 6, 8620–8631 CrossRef CAS PubMed.
- I. R. de Figueiredo, J. M. Freire, L. Flores, A. S. Veiga and M. A. R. B. Castanho, IUBMB Life, 2014, 66, 182–194 CrossRef PubMed.
- S. Deshayes, M. Morris, F. Heitz and G. Divita, Adv. Drug Delivery Rev., 2008, 60, 537–547 CrossRef CAS PubMed.
- P. Arukuusk, L. Pärnaste, H. Margus, N. K. Eriksson, L. Vasconcelos, K. Padari, M. Pooga and Ü. Langel, Bioconjugate Chem., 2013, 24, 1721–1732 CrossRef CAS PubMed.
- J. M. Williford, J. Wu, Y. Ren, M. M. Archang, K. W. Leong and H. Q. Mao, Annu. Rev. Biomed. Eng., 2014, 16, 347–370 CrossRef CAS PubMed.
- S. A. Moschos, S. W. Jones, M. M. Perry, A. E. Williams, J. S. Erjefalt, J. J. Turner, P. J. Barnes, B. S. Sproat, M. J. Gait and M. A. Lindsay, Bioconjugate Chem., 2007, 18, 1450–1459 CrossRef CAS PubMed.
- M. Jafari, W. Xu, S. Naahidi, B. Chen and P. Chen, J. Phys. Chem. B, 2012, 116, 13183–13191 CrossRef CAS PubMed.
- N. Ferrer-Miralles, J. L. Corchero, P. Kumar, J. A. Cedano, K. C. Gupta, A. Villaverde and E. Vazquez, Microb. Cell Fact., 2011, 10, 101 CrossRef CAS PubMed.
- X. L. Wang, S. Ramusovic, T. Nguyen and Z. R. Lu, Bioconjugate Chem., 2007, 18, 2169–2177 CrossRef CAS PubMed.
- L. Crombez, G. Aldrian-Herrada, K. Konate, Q. N. Nguyen, G. K. McMaster, R. Brasseur, F. Heitz and G. Divita, Mol. Ther., 2009, 17, 95–103 CrossRef CAS PubMed.
- K. Freimann, P. Arukuusk, K. Kurrikoff, L. D. F. Vasconcelos, K. L. Veiman, J. Uusna, H. Margus, A. T. Garcia-Sosa, M. Pooga and Ü. Langel, J. Controlled Release, 2016, 241, 135–143 CrossRef CAS PubMed.
- Y. Kato, S. Ozawa, C. Miyamoto, Y. Maehata, A. Suzuki, T. Maeda and Y. Baba, Cancer Cell Int., 2013, 13, 89 CrossRef CAS PubMed.
- S. Kimura, T. Kawano and T. Iwasaki, Biosci., Biotechnol., Biochem., 2017, 81, 112–118 CrossRef CAS PubMed.
- G. Moulay, C. Leborgne, A. J. Mason, C. Aisenbrey, A. Kichler and B. Bechinger, J. Pept. Sci., 2017, 23, 320–328 CrossRef CAS PubMed.
- Z. Meng, L. Luan, Z. Kang, S. Feng, Q. Meng and K. Liu, J. Mater. Chem., 2017, 5, 74–84 RSC.
- V. Iacobucci, F. Di Giuseppe, T. T. Bui, L. S. Vermeer, J. Patel, D. Scherman, A. Kichler, A. F. Drake and A. J. Mason, Biochim. Biophys. Acta, 2012, 1818, 1332–1341 CrossRef CAS PubMed.
- L. Prongidi-Fix, M. Sugawara, P. Bertani, J. Raya, C. Leborgne, A. Kichler and B. Bechinger, Biochemistry, 2007, 46, 11253–11262 CrossRef CAS PubMed.
- L. Pärnaste, P. Arukuusk, K. Langel, T. Tenson and Ü. Langel, Mol. Ther.–Nucleic Acids, 2017, 7, 1–10 CrossRef PubMed.
- Q. Leng, P. Scaria, J. Zhu, N. Ambulos, P. Campbell and A. J. Mixson, J. Gene Med., 2005, 7, 977–986 CrossRef CAS PubMed.
- W. Zhang, J. Song, B. Zhang, L. Liu, K. Wang and R. Wang, Bioconjugate Chem., 2011, 22, 1410–1415 CrossRef CAS PubMed.
- Q. Zhang, J. Tang, L. Fu, R. Ran, Y. Liu, M. Yuan and Q. He, Biomaterials, 2013, 34, 7980–7993 CrossRef CAS PubMed.
- Y. Zhang, L. Li, L. Chang, H. Liu, J. Song, Y. Liu, H. Bao, B. Liu, R. Wang and J. Ni, Chem. Biol. Drug Des., 2019 DOI:10.1111/cbdd.13537.
- O. Rötzschke, J. M. Lau, M. Hofstätter, K. Falk and J. L. Strominger, Proc. Natl. Acad. Sci. U. S. A., 2002, 26, 16946–16950 CrossRef PubMed.
- B. Chen, R. Pan, D. Askhatova and P. Chen, Int. J. Nanomed., 2015, 10, 3303–3314 CrossRef CAS PubMed.
- J. Regberg, L. Vasconcelos, F. Madani, Ü. Langel and M. Hällbrink, Int. J. Pharm., 2016, 501, 32–38 CrossRef CAS PubMed.
- T. Lehto, L. Vasconcelos, H. Margus, R. Figueroa, M. Pooga, M. Hällbrink and Ü. Langel, Bioconjugate Chem., 2017, 28, 782–792 CrossRef CAS PubMed.
- H. Margus, P. Arukuusk, Ü. Langel and M. Pooga, Mol. Pharm., 2016, 13, 172–179 CrossRef CAS PubMed.
- H. Margus, K. Padari and M. Pooga, Mol. Ther., 2012, 20, 525–533 CrossRef CAS PubMed.
- L. Shang, K. Nienhaus and G. U. Nienhaus, J. Nanobiotechnol., 2014, 12, 5 CrossRef PubMed.
- S. Bhattacharjee, J. Controlled Release, 2016, 235, 337–351 CrossRef CAS PubMed.
- C. Scholz and E. Wagner, J. Controlled Release, 2012, 161, 554–565 CrossRef CAS PubMed.
- P. J. Lin, Y. Y. Tam, I. Hafez, A. Sandhu, S. Chen, M. A. Ciufolini, I. R. Nabi and P. R. Cullis, Nanomedicine, 2013, 9, 233–246 CrossRef CAS PubMed.
- L. Gentilucci, R. De Marco and L. Cerisoli, Curr. Pharm. Des., 2010, 16, 3185–3203 CrossRef CAS PubMed.
- I. V. Chernikov, V. V. Vlassov and E. L. Chernolovskaya, Front. Pharmacol., 2019, 10, 444 CrossRef PubMed.
- B. C. Evans, C. E. Nelson, S. S. Yu, K. R. Beavers, A. J. Kim, H. Li, H. M. Nelson, T. D. Giorgio and C. L. Duvall, J. Visualized Exp., 2013, e50166 Search PubMed.
- D. Ma, S. Tian, J. Baryza, J. C. Luft and J. M. DeSimone, Mol. Pharm., 2015, 12, 3518–3526 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9bm00688e |
| This journal is © The Royal Society of Chemistry 2019 |