
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Amphiphilic tri- and tetra-block co-polymers combining versatile functionality with facile assembly into cytocompatible nanoparticles†
Catherine E.
Vasey‡
a,
Amanda K.
Pearce‡
 a,
Federica
Sodano
b,
Robert
Cavanagh
a,
Thais
Abelha
a,
Valentina
Cuzzucoli Crucitti
c,
Akosua B.
Anane-Adjei
a,
Marianne
Ashford
d,
Paul
Gellert
d,
Vincenzo
Taresco
*a and
Cameron
Alexander
*a
a,
Federica
Sodano
b,
Robert
Cavanagh
a,
Thais
Abelha
a,
Valentina
Cuzzucoli Crucitti
c,
Akosua B.
Anane-Adjei
a,
Marianne
Ashford
d,
Paul
Gellert
d,
Vincenzo
Taresco
*a and
Cameron
Alexander
*a
aSchool of Pharmacy, University Park University of Nottingham, NG7 2RD, UK. E-mail: vincenzo.taresco@nottingham.ac.uk; cameron.alexander@nottingham.ac.uk
bDepartment of Science and Drug Technology, University of Torino, 10125, Torino, Italy
cSchool of Chemical Engineering, University Park University of Nottingham, NG7 2RD, UK
dAstraZeneca, Pharmaceutical Sciences, Innovative Medicines, Silk Court Business Park, Macclesfield, Cheshire SK10 2NA, UK
First published on 9th July 2019
Abstract
In order for synthetic polymers to find widespread practical application as biomaterials, their syntheses must be easy to perform, utilising freely available building blocks, and should generate products which have no adverse effects on cells or tissue. In addition, it is highly desirable that the synthesis platform for the biomaterials can be adapted to generate polymers with a range of physical properties and macromolecular architectures, and with multiple functional handles to allow derivatisation with ‘actives’ for sensing or therapy. Here we describe the syntheses of amphiphilic tri- and tetra-block copolymers, using diazabicyclo[5.4.0]undec-5-ene (DBU) as a metal-free catalyst for ring-opening polymerisations of the widely-utilised monomer lactide combined with a functionalised protected cyclic carbonate. These syntheses employed PEGylated macroinitiators with varying chain lengths and architectures, as well as a labile-ester methacrylate initiator, and produced block copolymers with good control over monomer incorporation, molar masses, side-chain and terminal functionality and physico-chemical properties. Regardless of the nature of the initiators, the fidelity of the hydroxyl end group was maintained as confirmed by a second ROP chain extension step, and polymers with acryloyl/methacryloyl termini were able to undergo a second tandem reaction step, in particular thiol–ene click and RAFT polymerisations for the production of hyperbranched materials. Furthermore, the polymer side-chain functionalities could be easily deprotected to yield an active amine which could be subsequently coupled to a drug molecule in good yields. The resultant amphiphilic copolymers formed a range of unimolecular or kinetically-trapped micellar-like nanoparticles in aqueous environments, and the non-cationic polymers were all well-tolerated by MCF-7 breast cancer cells. The rapid and facile route to such highly adaptable polymers, as demonstrated here, offers promise for a range of bio materials applications.
Introduction
Biodegradable synthetic polymer–drug conjugates have been under active investigation as macromolecular therapeutics for over 40 years, yet there are still relatively few marketed drugs of this type.1–4 One major reason for this lack of commercial and clinical translation has been the difficulty in matching the conflicting demands of high therapeutic efficacy with ease of preparation and characterisation. For example, there have been many examples of synthetic polymer–drug conjugates which show high activity in vitro and in pre-clinical in vivo studies.5–8 However, inspection of the structures of some of these materials suggests they would be challenging to synthesise from a commercial standpoint, or would require extensive toxicological investigations prior to use which would slow their development.9,10 Accordingly, there remains a focus on aliphatic polyesters for pharmaceutical applications, as there are many flexible routes for their synthesis, and the metabolites produced upon their breakdown in the body are largely known and are removed via natural excretory pathways.11–13 Poly(lactide)s (PDLLA), poly(caprolactone)s (PCL) and poly(glycolide)s (PGA) and their copolymers are the most widely used polyesters in the health care sector, owing to their accessibility from readily-available monomers, favourable mechanical properties, hydrolytic degradation and biocompatibility.14–17Of particular interest are poly(lactide)s and associated copolymers which can be easily synthesised via ROP of D,L-lactide (LA), a natural recurring cyclic ester. PDLLA is already used in drug delivery as the hydrophobic block of GenexolP10®, a PEG-PLA co-polymer formulation of the anti-cancer drug Paclitaxel,18 currently in the clinic in Korea. However, a major drawback with lactide-type polymers in advanced therapeutic applications is the lack of functional ‘handles’ on the monomer units, resulting in the inability to conjugate drug molecules to the polymer backbone. This limits the overall drug loading in the formulation and ultimately the dose which can be delivered. In addition, limitations in the ability to control degradation rate, as well as concerns over the long-term biological effects of tin-based catalysts typically used in their production,19,20 are driving research efforts towards the development of more ‘functional’ and adaptable PLA-analogue polymer therapeutics.
Modification of the starting lactide monomers can be employed to introduce addressable functionalities along the polymer main chain.21 However, this strategy often requires many synthetic steps as well as inefficient purifications leading to low yields. Another disadvantage is that not all introduced functionalities are compatible with the ROP process. Free nucleophiles such as hydroxyls, amines or thiols cannot be present during polymerisation, and therefore protection/deprotection and post-polymerisation modifications may need to be employed if drugs are to be linked to the polymers. In this regard, aliphatic poly(carbonate)s are advantageous, as there are a range of methods for their derivatisation, and the central polymer backbone has appropriate properties for biomedical applications, being well-tolerated in the body and also biodegradable.22 In addition, main-chain substituted polycarbonates can be synthesised from functionalised cyclic carbonate monomers through relatively simple chemistry, thus allowing ready access to covalently-linked polymer–drug conjugates.23
There are now several reports that both lactide and cyclic carbonate monomers can be ring-opened under ambient conditions with organocatalysts, such as 1,8-diazabicyclo[5.4.0]undec-5-ene (DBU),24–27 thus negating the need for metal catalysts or high temperatures which might otherwise generate toxic or degraded components in the final polymer product. These data suggest that functionalised materials which are (a) biodegradable and (b) adaptable for a variety of therapeutic formats might be produced through ring-opening polymerisations of carbonates under mild reaction conditions. These chemistries might also serve to generate ‘platform’ polymer–drug conjugates which can be optimised for specific drug delivery applications by further end-group or side-chain modifications. For example, by utilising PEG chains of varying lengths and architectures as initiators, amphiphilic PEG-poly(caprolactone) co-polymers which self-assemble into long-circulating nanoparticles can be envisaged. Importantly, the adoption of bifunctional initiators provides additional opportunities to produce biodegradable copolymers with hybrid architectures by using the ω-functionality in a second synthetic step, such as ATRP or RAFT polymerisations, or Michael addition reactions.28
In this work we report the synthesis of a small library of mixed-polyester–polycarbonate materials with potential applications in polymer therapeutics (Fig. 1).
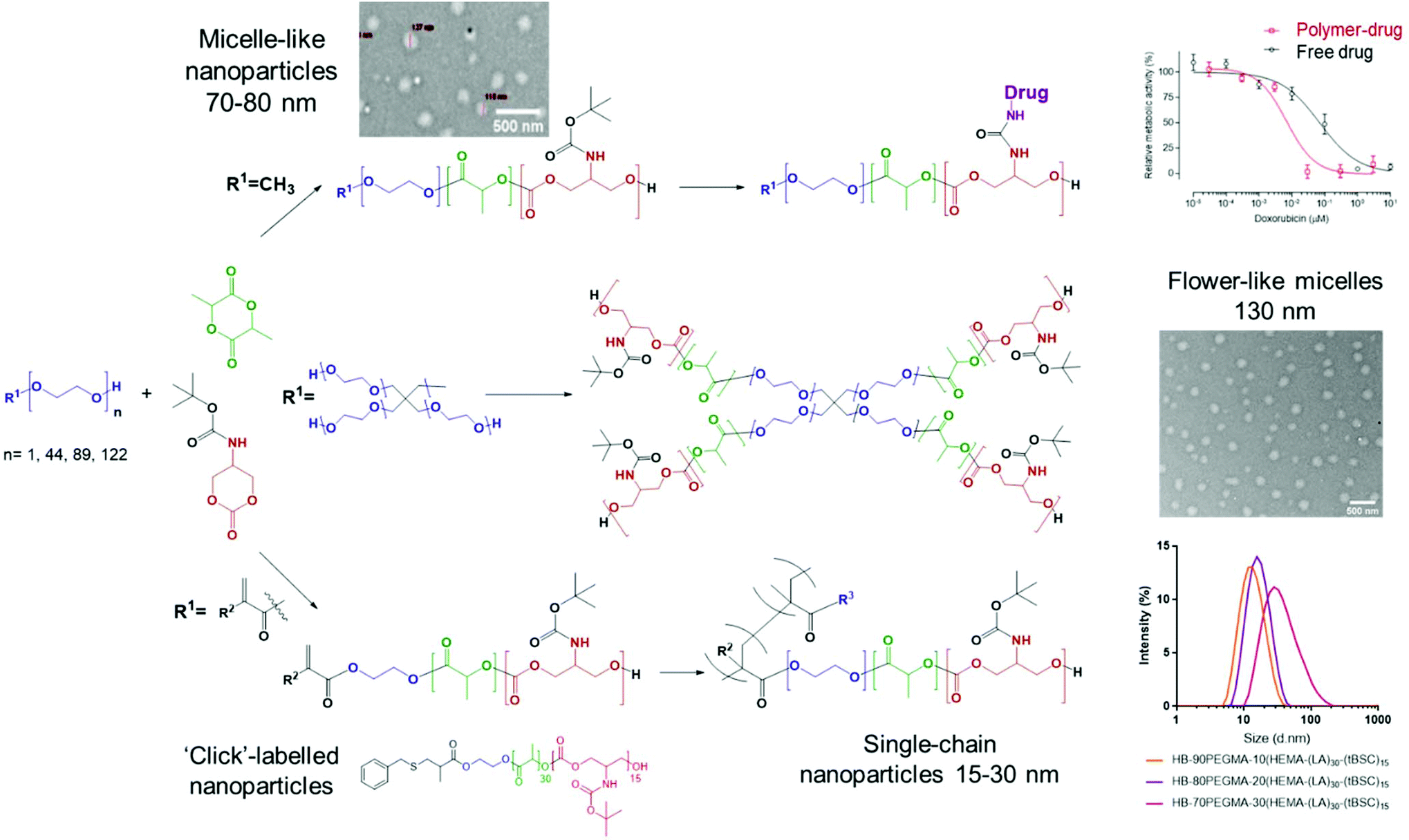 | ||
| Fig. 1 Synthetic strategy towards multicomponent amphiphilic copolymers and key characterisation outputs. | ||
The route involved preparation of a BOC-protected cyclic carbonate monomer (tBSC) derived from serinol, followed by ROP with lactide, initiated by a range of nucleophiles, to generate the multi-architecture functional materials. The different PEG-based initiators were chosen to tune the amphiphilic balance of the final block-copolymer chain (keeping the hydrophobic block length constant), in order to produce polymers capable of self-assembly into nanoparticles with a range of sizes. To highlight further the versatility of the approach, a methacrylate initiator was employed to generate an end-group used in tandem ROP-thiol–ene “click” and ROP-RAFT polymerisations, thus forming amphiphilic hyperbranched polymers with different sizes. In addition, after deprotection of the BOC groups on the polycarbonate component of the materials, the resultant amine functionality was exploited to link with doxorubicin via a urea group as a proof of concept for drug conjugation. Finally, the polymer library was evaluated for effects on a model cell line as the first steps towards practical drug delivery applications.
Experimental section
Materials
All chemicals were used as obtained without additional purification unless otherwise stated. Poly(ethylene glycol) methyl ether (Mn = 5000/2000 Da), trimethylolpropane ethoxylate (3PEG, 4/15 EO/OH, Mn = 1014 Da), pentaerythritol ethoxylate (4PEG, 15/4 EO/OH, Mn = 797 Da), 1,8-diazabicyclo[5.4.0]undec-5-ene (DBU, 98%), triethylamine (TEA, ≥99%), 4-aminofluorescein, di-tert-butyldicarbonate (99%), 4-(hydroxymethyl)phenylboronic acid pinacol ester (97%), and extra-dry DMSO were purchased from Sigma-Aldrich. Polyethylene glycol (PEG, average Mn = 4000 Da) was purchased from Fisher Chemical. 2-Amino-1,3-propanediol (Serinol, 98%) and dichloromethane (DCM, 99.8%, extra dry over molecular sieve, stabilised, Acroseal) were purchased from AcrosOrganics. Trifluoroacetic acid (TFA, 99%) was purchased from VWR Chemicals. Ethylchloroformate was purchased from Merck KGaA.Synthesis of tert-butyl (1,3-dihydroxypropan-2yl) carbamate (tert-butyl serinol, tBS)
Serinol (10 g, 110 mmol) was dissolved in a mixture of water and tetrahydrofuran (THF) (100 ml, 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) and the solution was cooled to 0 °C. Following addition of triethylamine (22.5 mL, 162 mmol), to this mixture di-tert-butyl dicarbonate (29 g, 131.5 mmol) pre-dissolved in THF (20 mL) was added dropwise over 1 h. The reaction mixture was left stirring overnight at room temperature. Subsequently, THF was removed under reduced pressure and the residue was extracted with ethyl acetate (4 × 100 mL). The colourless organic phase was dried over anhydrous magnesium sulphate and concentrated under reduced pressure. The crude product was recrystallized in ethyl acetate (yield 70%, 14.7 g). 1H NMR (400 MHz, CDCl3, ppm): δ 5.30 (s, 1H), 3.86–3.63 (m, 5H), 2.60 (s, 2H), 1.45 (s, 12H). Observed chemical shifts and integrals were in agreement with previous reports.29
:1 v/v) and the solution was cooled to 0 °C. Following addition of triethylamine (22.5 mL, 162 mmol), to this mixture di-tert-butyl dicarbonate (29 g, 131.5 mmol) pre-dissolved in THF (20 mL) was added dropwise over 1 h. The reaction mixture was left stirring overnight at room temperature. Subsequently, THF was removed under reduced pressure and the residue was extracted with ethyl acetate (4 × 100 mL). The colourless organic phase was dried over anhydrous magnesium sulphate and concentrated under reduced pressure. The crude product was recrystallized in ethyl acetate (yield 70%, 14.7 g). 1H NMR (400 MHz, CDCl3, ppm): δ 5.30 (s, 1H), 3.86–3.63 (m, 5H), 2.60 (s, 2H), 1.45 (s, 12H). Observed chemical shifts and integrals were in agreement with previous reports.29
Synthesis of tert-butyl (2-oxo-1,3-dioxan-5-yl) carbamate: (tert-butyl serinol carbonate, tBSC)
Tert-Butyl (1,3-dihydroxypropan-2yl) carbamate (26.0 g, 136 mmol) and ethyl chloroformate (32.0 g, 295 mmol) were dissolved in THF (140 mL) and the solution was cooled to 0 °C. Triethylamine (47.0 mL, 338 mmol) was then added dropwise over 1 h. Subsequently, the reaction mixture was left stirring overnight at room temperature. The precipitated salts were filtered off and the filtrate was concentrated under reduced pressure to afford the product as a white solid. The product was left drying in a vacuum oven overnight and subsequently washed with a mixture of THF:Et2O (2:8, v/v) to remove traces of unreacted starting materials (yield 65%, 19.20 g). 1H NMR (400 MHz, CDCl3, ppm): δ 5.30 (s, 1H), 4.55 (d, 2H), 4.45 (d, 2H), 4.10 (s, 1H), 1.45 (s, 9H). Observed chemical shifts and integrals were in agreement with previous reports.29,30
PEGylated initiated ring-opening polymerisation of lactide and tert-butyl (2-oxo-1,3-dioxan-5-yl) carbamate (tBSC): kinetics in presence of DBU
A predetermined amount of PEGylated chain initiator (mPEG2000, mPEG5000, PEG4000, tPEG1014 or pPEG797), lactide and tBSC were weighed out in a glass vial (dried in an oven at 100 °C overnight) and capped with a rubber stopper. [M]/[I] was adjusted in order to maintain a constant length of hydrophobic chains across the different final architectures. Dry DCM (5 mL), was added and the mixture was allowed to dissolve at room temperature. An aliquot was taken of the monomer–initiator mixture at this point: this sample was denoted as at time point t0. DBU (3% mol/mol compared to the total amount of monomers) was dissolved in DCM (1 mL) and added to catalyse the ring opening process. After reaction, the catalyst was deactivated through adding the polymer mixture, dropwise, to cold hexane (30 mL) and the polymer was purified via multiple precipitation steps and dried in a vacuum oven (conversion of monomer into polymer ∼75–80% w/w). The purified materials were analysed by NMR and GPC. 1H NMR (mPEG5000-(LA)50-(tBSC)50 as model) (400 MHz, CDCl3, ppm): δ 5.19 (broad m, 104H), 4.26 (broad m, 249H), 3.66 (broad s, 492H), 3.39 (s, 3H), 1.82–1.46 (2 asymmetric broad s, 429 + 35H), 1.59 (broad m, 314H). 13C NMR (101 MHz, CDCl3, ppm) δ = 169.76, 154.50, 70.57, 69.00, 67.97, 65.58, 30.93, 28.33, 25.61, 16.65, 15.28.PEGylated initiated ring-opening polymerisation of sequential block copolymer of lactide and tert-butyl (2-oxo-1,3-dioxan-5-yl) carbamate (tBSC): kinetics in presence of DBU
A predetermined amount of PEGylated chain initiator (PEG4000) and lactide were weighed out in a glass vial (dried in an oven at 100 °C overnight) and capped with a rubber stopper. Dry DCM (5 mL), was added and the mixture was allowed to dissolve at room temperature. Aliquots were taken and polymerisation carried out as described for the mPEG-OH reaction. After polymerisation, the catalyst was deactivated through adding the polymer mixture, dropwise, to cold hexane (30 mL) and the polymer was purified via multiple precipitation steps and dried in a vacuum oven to obtain PEG4000-(LA)50 (P6). The resultant macroinitiator and (tBSC) was weighed out in a glass vial, and the polymerisation carried out as above to achieve the chain extension. After 15 minutes of reaction time, the catalyst was deactivated through adding the polymer mixture, dropwise, to cold hexane (30 mL) and the polymer was purified and dried as before to obtain PEG4000-(LA)50-b-(tBSC)15 (P7) (conversion of monomer to polymer ∼75–80% w/w). A final NMR spectrum was recorded of the products post purification. 1H NMR (400 MHz, CDCl3, ppm): δ 5.19 (broad m, 94H), 4.28 (broad m, 68H), 3.67 (broad s, 356H), 1.65–1.47 (2 asymmetric broad s53 + 78H), 1.61 (broad m, 280H). 13C NMR (101 MHz, CDCl3, ppm) δ = 169.53, 154.52, 70.57, 69.02, 66.03, 31.22, 16.66.(2-Hydroxyethyl)methacrylate initiated ROP of lactide and (tBSC) in presence of DBU
The desired amounts of cyclic monomers and HEMA-initiator were weighed into a vial (dried in an oven at 100 °C overnight). The initiator was stored under anhydrous conditions. Subsequently, the vial was capped using a rubber stopper, and DCM, dried over molecular sieves and kept under an inert gas environment, was added and the mixture was allowed to dissolve properly (at RT) for 5–10 minutes. The catalyst was added at 1.5% mol/mol compared to the amount of monomer to trigger the ring opening process. After 15 minutes of reaction time, the catalyst was deactivated through adding the polymer mixture, dropwise, to cold diethyl ether (20 mL) and the polymer was purified via multiple precipitation steps and dried in a vacuum oven to obtain the final macromonomer (conversion of monomer into polymer circa 75–80% w/w). A final NMR spectrum was recorded of the product post purification.Thiol–ene “click” chemistry of macromonomers
HEMA-(LA)15-(tBSC)15 (P8) (15 mg) was dissolved in 30 μL of DMSO-d6. 2,2-Dimethyl-2-phenyl acetophenone (DMPA) (25 μL aliquot of a 40 mg mL−1 solution in DMSO-d6) and benzyl mercaptan (5 equiv.) were added and the reactions were then irradiated by UV at 365 nm for 30 minutes. The modified macromonomer was purified by precipitation into cold diethyl ether (5 mL) and dried. A final NMR spectrum was recorded of the product post purification.RAFT polymerisation of macromonomers
BOC deprotection optimisation
A predetermined amount of PEGylated copolymer (P2, 100 mg, 0.46 mmol in carbonate-BOC monomer) was dissolved in extra dry DCM (3 mL). The TFA optimum amount was found to be 5 eq. with respect to the calculated tBSC units (2.3 mmol, 263 mg) and was added slowly to the polymer mixture at 0 °C. The reaction mixture was left stirring for varying amounts of time (5, 10 and 30 min) and following precipitation in cold hexane, was washed with cold diethyl ether and analysed by 1H-NMR. Different degrees of deprotection were achieved by simply varying the reaction time and at the same time polymer main backbone degradation was avoided. 1H NMR (t = 0 min) (400 MHz, DMSO-d6) δ = 5.26–5.10 (m, 80H), 4.17–4.01 (m, 168H), 3.52 (s, 492H), 1.48 (s, 240H), 1.39 (s, 370H). 1H NMR (after 30 min and post purification and drying) (400 MHz, DMSO-d6) δ = 8.42 (bs, 30H), 7.18–7.02 (m, 34H), [around 10 units deprotected] 5.24–5.11 (m, 62H), 3.51 (s, 492H), 1.55–1.41 (m, 204H), 1.38 (s, 320H).Doxorubicin coupling procedure
N,N′-Disuccinimidyl carbonate (DSC) (1.5 eq. with respect to the amount of doxorubicin, 0.125 mmol, 32 mg) pre-dissolved in anhydrous MeCN (3 mL) was added to a solution of doxorubicin (1.0 eq., 0.083 mmol, 48 mg) in dry DMSO (3 mL) at 0 °C. TEA (2.5 eq. with respect to the amount of doxorubicin, 0.21 mmol, 30 μL) was added slowly to the reaction mixture and left stirring for 4–5 hours. Deprotected polymer (mPEG5000-(LA)50-(tBSC)50; 1.0 eq., 0.083 mmol, 15 mg; equivalents were calculated to be in high excess when compared to each repeating tBSC unit) was dissolved in a mixture of anhydrous MeCN/dry DMSO (50/50, v/v, 3 mL) and TEA (2.0 eq. with respect to the amount of deprotected polymer, 0.166 mmol, 23 μL) was added, and the solution was subsequently added dropwise to the previous reaction mixture. The reaction was left stirring for a further 3 hours at 0 °C. The resultant conjugated polymer was purified through multiple precipitation steps in cold hexane:diethyl ether (1:5 v/v) mixture and dialysed against water:methanol (5:1 v/v) mixture for 24 h. Finally, the purified aqueous suspension was freeze-dried for 48 h and stored at −22 °C. A final NMR spectrum of the product (mPEG5000-(LA)50-(tBSC)50-DOXO) post purification and completely dried was recorded. 1H NMR (400 MHz, DMSO-d6) δ = 14.08 (s, 4H), 7.93 (d, J = 4.3 Hz, 8H), 7.69–7.65 (m, 4H), 7.63 (s, 4H), 7.07 (d, J = 7.1 Hz, 21H), 5.34 (d, J = 8.7 Hz, 8H), 5.24–5.11 (m, 78H), 4.86 (d, J = 5.9 Hz, 4H), 4.61–4.56 (m, 4H), 4.50 (d, J = 10.1 Hz, 8H), 4.36 (d, J = 4.0 Hz, 12H), 4.16–3.93 (m, 205H), 3.81–3.74 (m, 8H), 3.71–3.66 (m, 4H), 3.51 (s, 492H), 3.24 (s, 4H), 3.07–3.01 (m, 8H), 2.94 (d, J = 5.9 Hz, 4H), 2.23 (d, J = 3.8 Hz, 4H), 1.50–1.41 (m, 200H), 1.38 (s, 290H).
Nanoparticle preparation
A nanoprecipitation method was used to form nanoparticles, whereby the polymers (10 mg) were dissolved in acetone (1 mL). The polymeric solution was added dropwise to deionized water (10 mL, final concentration of 1 mg mL−1), under constant stirring at 550 rpm. The polymers rapidly formed nanoparticle suspensions through solvent exchange between water and acetone. The final suspension was then left stirring overnight at room temperature in order to reach complete acetone evaporation.Critical aggregation concentration (CAC) calculation
The critical aggregation concentration (CAC) was calculated using a Zetasizer Nano ZS as described in Malvern application note AN101104 (http://www.malvern.com/en/support/resource-center/application-notes/AN101104SurfactantMicelleCharacterization.aspx). In brief, a constant attenuator was selected and the intensity of the scattered light (count rate in kcps) was monitored for a range of diluted NP suspensions (100–0.05 μg mL−1).31 The intensity increases linearly with concentration above the CMC, while below the CMC the count rates reach a plateau. The CMC was then calculated by intersecting the two lines.4-Aminofluorescein encapsulation study
Polymer (10 mg) was dissolved in acetone (1 mL). An aliquot (0.5 mL) of a stock solution of 4-aminofluorescein (5.27 mg dissolved in 3.30 mL acetone) was added to the polymer. This was then added dropwise to PBS (10 mL) under constant stirring at 550 rpm. After 3 hours, nanoparticle colloidal suspensions were left to dialyse in water for 4 days.HB-PEGylated copolymers coprecipitation
Polymers P2 or P5 (10 mg) were dissolved in acetone (1 mL) and polymer HB2 (10 mg) was added to the organic solution. The final solutions containing both the PEGylated copolymers and HB materials were then added dropwise to PBS (10 mL) under constant stirring at 550 rpm. After complete acetone evaporation DLS analysis was performed to evaluate the aggregation state of the final colloidal systems. As a control, the two suspensions (copolymers alone and HB alone) prepared in two separate precipitations steps were mixed directly in the DLS cuvette (0.5 mL each).Effects of polymer formulations in breast cancer cells
The lactate dehydrogenase (LDH) release assay and PrestoBlue™ Cell Viability assays were performed to assess the effects of nanoparticles on a model cell line. MCF7 breast cancer cell line was obtained from American Type Culture Collection (ATCC) and cultured in passage window of 15. Cells were cultured in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% Fetal Bovine Serum (FBS), 0.1 mg ml−1 streptomycin, 100 units per ml penicillin, 0.25 μg ml−1 amphotericin and 2 mM L-glutamine. The cells were seeded at 1 × 104 cells per well in 96 well plates and cultured for 24 hours prior to assaying. Nanoparticles were exposed to cells for 4, 24 or 48 hours and applied in 100 μL phenol red free DMEM containing 10% (v/v) FBS and 2 mM L-glutamine. Triton X-100 applied at 1% (v/v) applied in phenol red free medium was used as a cell death (positive) control and a vehicle control containing no nanoparticles used as a negative control. Following exposure, 50 μL of supernatant was collected per well for analysis of LDH content. Cells were then washed twice with warm PBS and 100 μL 10% (v/v) PrestoBlue reagent diluted in phenol red free medium applied per well for 60 minutes. The resulting fluorescence was measured at 560/600 nm (λex./λem.). Relative metabolic activity was calculated by setting values from the negative control as 100% and positive control values as 0% metabolic activity. Assessment of LDH release was performed according to the manufacturer's instructions and involved adding 100 μL LDH detection reagent to the collected supernatant samples and incubating at room temperature shielded from light for 25 minutes. Absorbance was then measured at 492 nm. Relative LDH release was calculated with the negative control absorbance at 492 nm taken as 0%, and the positive control, assumed to cause total cell lysis, as 100%.General methods and instrumentation
800000–505 g mol−1) and polycaprolactone standards were used for calibration.
Results and discussion
The key steps in the syntheses were preparation of a side-chain functional cyclic carbonate monomer, and ring-opening polymerisations with organic catalysts under mild conditions to generate materials with a range of architectures and self-assembly properties.Monomer synthesis and purification
The cyclic carbonate monomer was prepared by adapting a previously reported two-step synthesis.32 The key modification was the removal of chromatographic purification steps in the monomer synthesis by a simple solvent/non solvent (THF/Et2O) wash step. The final unoptimised yield of pure monomer was 65% after this rapid and simplified synthetic route (Fig. S1†).Polymer syntheses – statistical co-polymers with polyester and polycarbonate hydrophobic blocks
Five different PEGylated initiators were adopted to produce polymers with different final architectures, but the same overall length of hydrophobic unit, which in this case was a random copolymer of lactide and tBSC. It was observed via NMR kinetics experiments that both monomers were consumed to >90% conversion within 15 minutes (Fig. S2†), and therefore this was chosen as the reaction time for all the PEG-initiated polymerisations. As depicted in Fig. 2, all of the final polymers showed the characteristic NMR peaks of polymerised lactide and tBSC, with good agreements in the feed ratios of both monomers and the initiator (Fig. S3,† mPEG5000-(LA)50-(tBSC)50 as model, P2 entry). In addition, in the ATR-IR spectra, all the characteristic transitions of both ester and carbonate moieties were observed (Fig. S4†), further confirming the successful copolymerisation. There was no observed difference in reactivity across the series of PEGylated initiators, as demonstrated by the final molecular weights and monomer ratios reached post-purification (Table 1) (Scheme 1).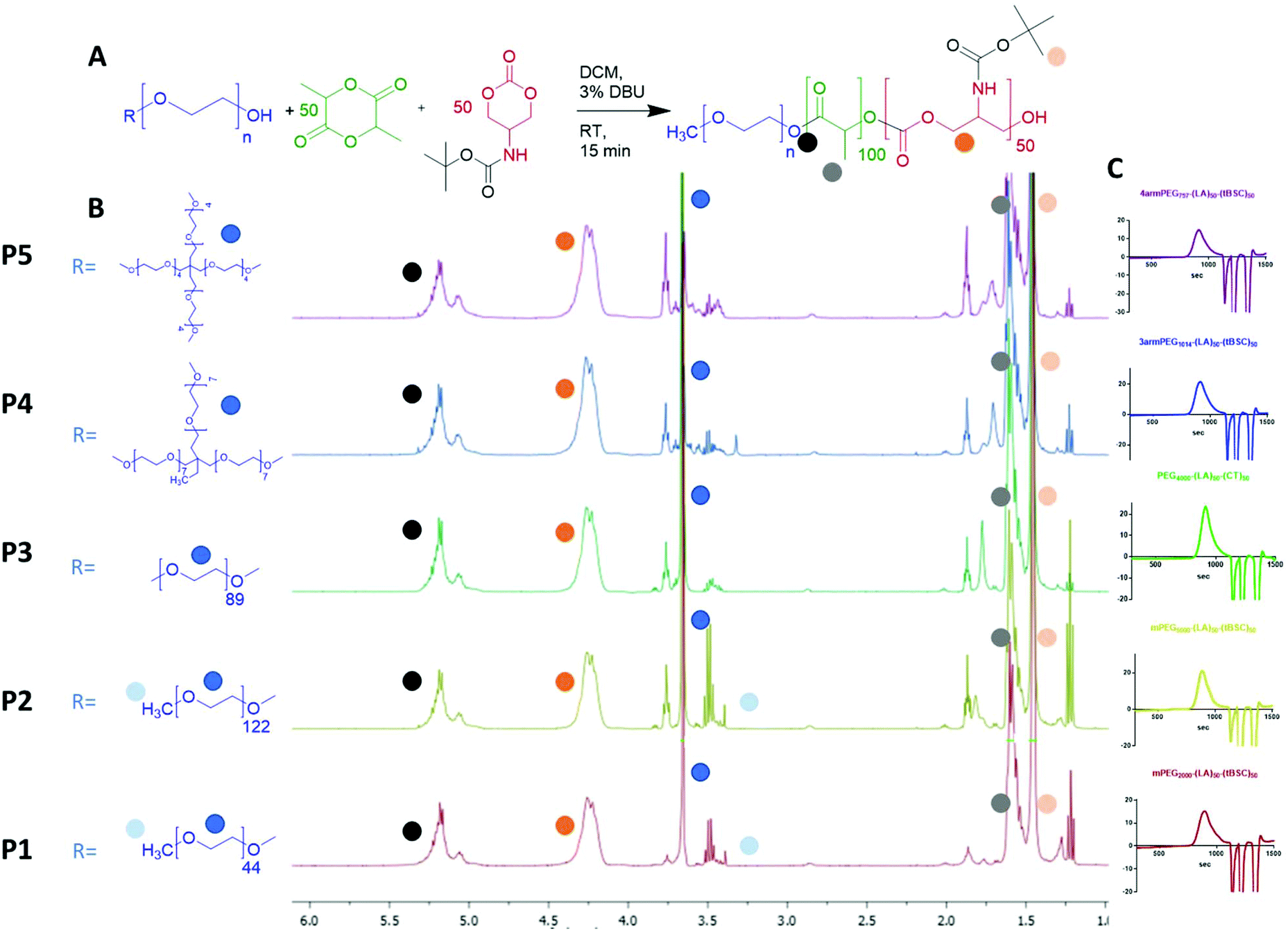 | ||
| Fig. 2 (A) general reaction scheme for the PEG-initiated ROP, (B) 1H NMR spectra of the five copolymers (with random distribution of ester and carbonate units) and (C) GPC traces of the polymers. | ||
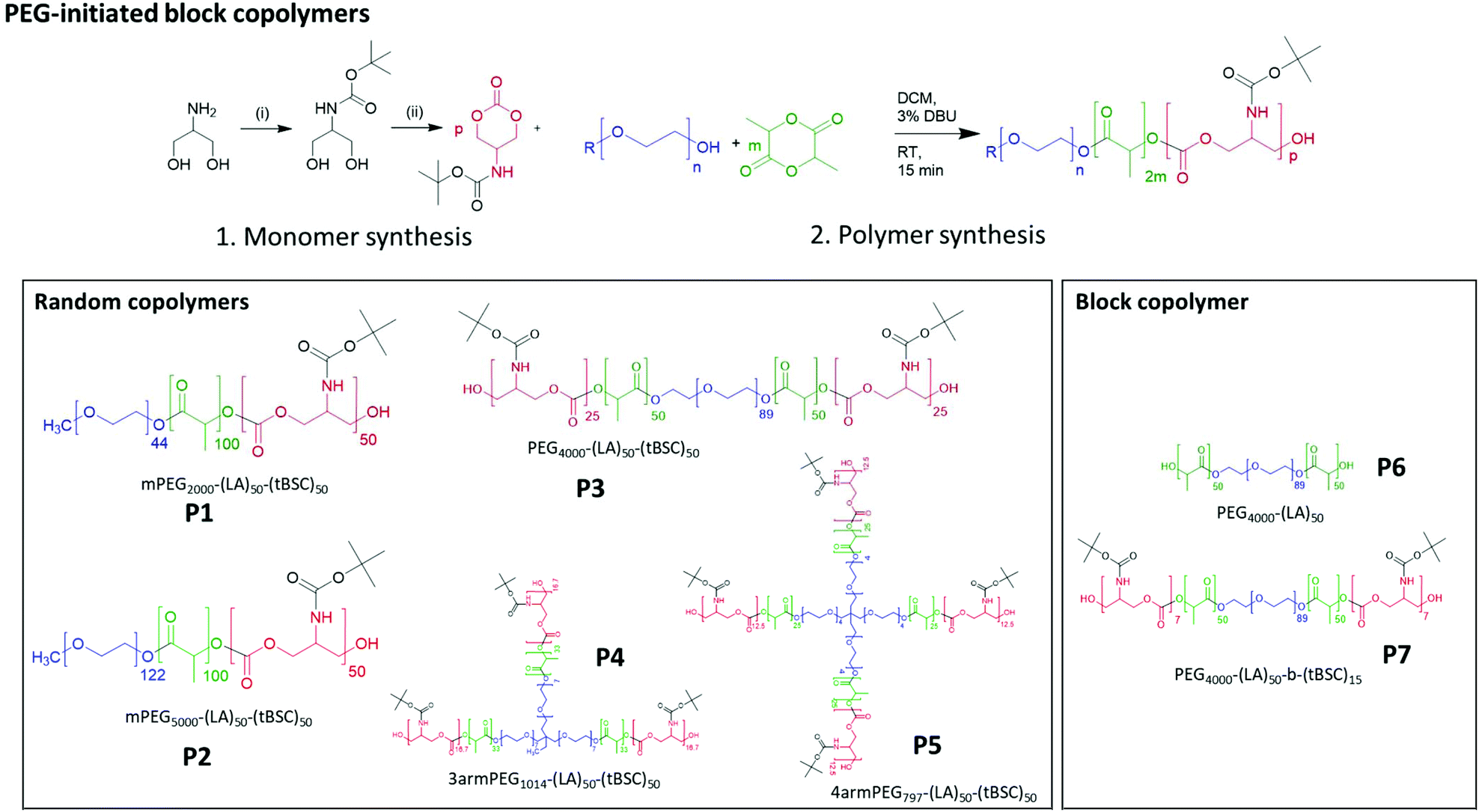 | ||
| Scheme 1 Synthesis of monomer and PEG-initiated copolymers of varying architectures and block compositions. | ||
| LA-tBSC units (targeted) | LA-tBSC units (exper) | M n (NMR) | M n (GPC) kDa | Đ | T g (°C) | T m1 (°C) | T m2 (°C) | ΔH1 (J g−1) | |
|---|---|---|---|---|---|---|---|---|---|
| a Compared to PMMA standards. | |||||||||
| P1. mPEG2000-(LA)50-(tBSC)50 | 60–53 | 55–48 | 20330 |
20.3 | 1.26 | 43.1 | 39.6 | — | 5.5 |
| P2. mPEG5000-(LA)50-(tBSC)50 | 55–50 | 52–50 | 23200 |
23.3 | 1.15 | 18.6 | 45.2 | — | 31.7 |
| P3. PEG4000-(LA)50-(tBSC)50 | 50–50 | 50–45 | 20960 |
17.0 | 1.22 | 20.6 | 42.1 | — | 25.6 |
| P4. 3 armPEG1014-(LA)50-(tBSC)50 | 55–53 | 52–49 | 19135 |
16.4 | 1.21 | 46.4 | — | — | — |
| P5. 4 armPEG797-(LA)50-(tBSC)50 | 55–55 | 55–55 | 20650 |
16.3 | 1.22 | 42.1 | — | — | — |
| P6. PEG4000-(LA)50 | 50–— | 50–— | 11200 |
10.2 | 1.05 | 42.2 | 49.2 | — | 21.0 |
| P7. PEG4000-(LA)50-b-(tBSC)15 | 50–15 | 50–13 | 14020 |
13.6 | 1.18 | n.d. | 41.2 | — | 26.2 |
In the DSC thermograms, as reported in Table 1, the polymers synthesised using crystalline PEG initiators showed a melting point (with different values of enthalpy) in the first cycle, while no melting transition could be observed in subsequent cooling and heating cycles within the temperature range explored. Glass transition temperatures (Tg) were observed for all the polymeric samples indicating the presence of amorphous structures as previously reported for similar copolymers such PEG-PLGA and PLGA.33,34
The inability of the PEG chain to recrystallise can be attributed to the interaction with the ester-carbonate hydrophobic chains, hindering the chain-packing nucleation steps during cooling cycles.35 In addition, it was found that the polymers synthesised from PEG initiators which were liquid at room temperature (i.e. the 3 and 4 arms PEG, P4 and P5 entries) had no determinable melting point. Furthermore, Thermal Gravimetric Analysis (TGA), demonstrated that the five copolymers were thermally stable up to 160–180 °C. Above this temperature, a steep loss in mass occurred up to 90% of the initial sample (P2 and P3 copolymer showed a loss around 70% attributed to a higher thermal stability), with the end of the transition at around 240–260 °C (Fig. S5†). During this thermal transition, two degradation steps can be observed (Fig. S5†) after the derivative analysis (Fig. S5† inset). These two thermal degradation transitions are likely to be related to lactide (∼30% weight loss) and carbonate (∼50% weight loss), respectively, due to the final amount in weight in the polymers. The higher weight retention of P2 and P3 copolymers has been attributed to the higher weight contribution of the PEG chains with a net initial weight degradation transition with an onset at above 300 °C, typical of longer PEG chains.36 The similar thermal degradation profiles of the five copolymers highlighted the similarity in properties of the main ester-carbonate fragment.
End group fidelity, block copolymer extension and hybrid polymer architectures via mixed methacrylate-polyester-co-polycarbonates
To confirm both the presence and fidelity of the hydroxyl end group following the polymerisations, as well as increase the range of polymeric architectures achieved, a chain extension reaction was carried out from a PEG4000-poly(lactide) polymer. Accordingly, 50 units of lactide as the first monomer were polymerised and the interim structure confirmed by NMR and GPC following purification (Fig. S6†). Subsequently, the resultant PEG-lactide macroinitiator (P6) was further reacted with tBSC to form the block copolymer (P7). The successful attainment of the A–B–C block was certified by NMR, GPC and ATR-IR of the final material, which demonstrated that no unanticipated side-reactions occurred during the DBU catalysed polymerisation (Fig. S6†). As expected, the chain extension of the PEG-lactide A–B polymer, P6, with the carbonate to form the A–B–C copolymer, P7, modified the thermal response of the final materials, as seen in the DSC thermograms, where the melting point of the first cycle shifted over 7 °C (from 49–42 °C) (Table 1). No melting transitions were observed for either P6 or P7 in the second thermal cycle.Further confirmation of end-group fidelity, but in this case from the initiating end of the polymer, was carried out by preparing a mixed ester-carbonate macromonomer using 2-hydroxyethylmethacrylate (HEMA) as the initiator (Scheme 2).
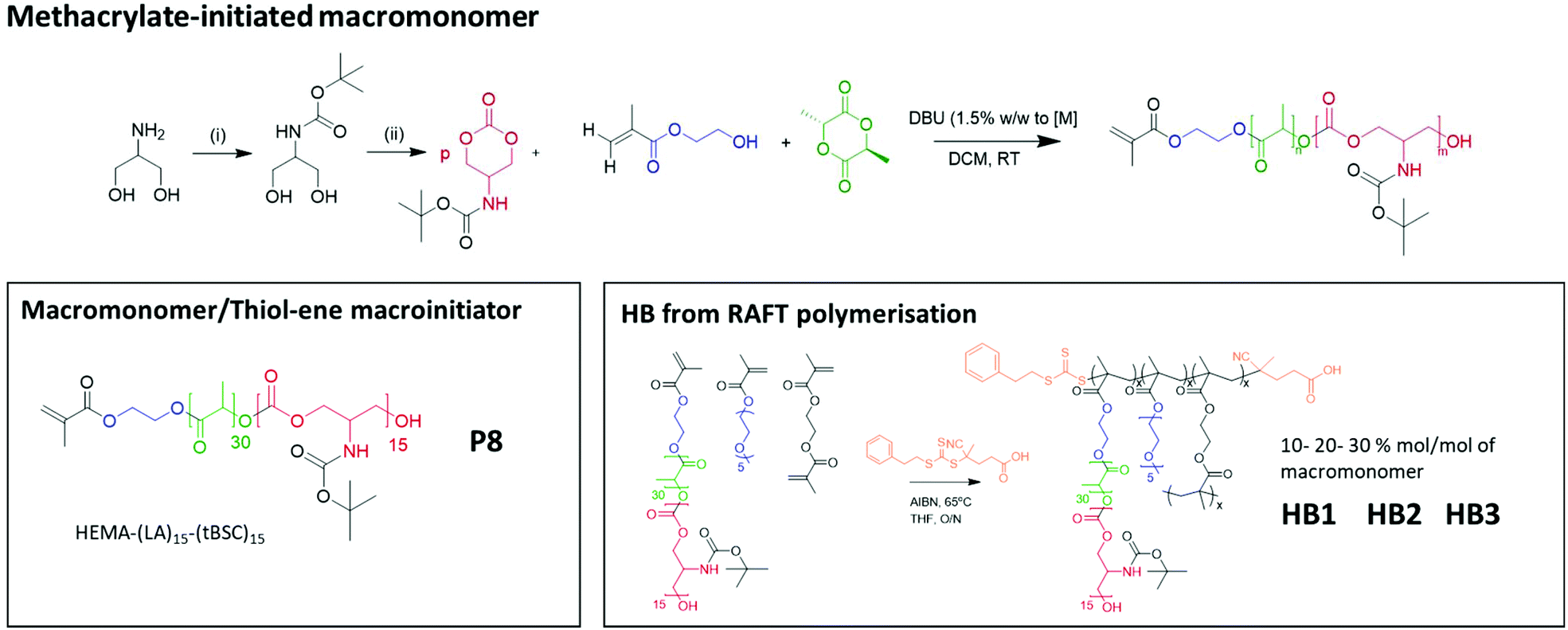 | ||
| Scheme 2 The overall reaction steps of the production of labile-ester initiated copolymer, from monomer synthesis to the formation of varying hyperbranched polymers. | ||
Therefore, HEMA was employed as an initiator to synthesise random mixed ester-carbonate macromonomers targeting final DPn values of 15 for LA and for tBSC. The reactions reached conversions of >90% within 15 minutes with no side-reactions as indicated by NMR (Fig. S7†). The expected retention of methacrylate functionality was demonstrated by a radical thiol–ene “click” reaction of the P8 macromonomer with benzyl mercaptan as the model thiol. The macromonomer was reacted with 5 equivalents of the thiol with DMPA as photoinitiator, followed by irradiation at 365 nm for 30 minutes.37,38
Complete conversion of the vinyl groups into the thioether product was achieved as confirmed by the 1H NMR spectra (Fig. S8†), marked by the disappearance of the vinyl protons (between 5.7–6.1 ppm) and the appearance of the diagnostic benzyl protons (7.2–7.4 ppm).
The extension of the mixed methacrylate polyester-carbonate platform into single-chain nanoparticles was subsequently investigated owing to the many potential applications of these systems in diagnostic and therapeutic applications.39–41 The HEMA-(LA)15-(tBSC)15 macromonomer was copolymerised with the hydrophilic monomer poly(ethyleneglycolmethacrylate) (PEGMA) in the presence of a bifunctional cross linker (ethyleneglycoldimethacrylate, EGDMA) at 10, 20 and 30 mol% with respect to PEGMA. The 10 and 20 mol% monomer composition resulted in branched particles, HB1 and HB2, that retained their aqueous solubility and with sizes of ∼13 nm and ∼15 nm respectively. However, the 30 mol% monomer composition was insoluble in water, and thus was further chain extended with additional PEGMA until the water-soluble polymer HB3 was produced, which displayed a final particle size of 30 nm. As for the linear polymers, GPC analysis confirmed a single polymeric population in all cases (Fig. 3).
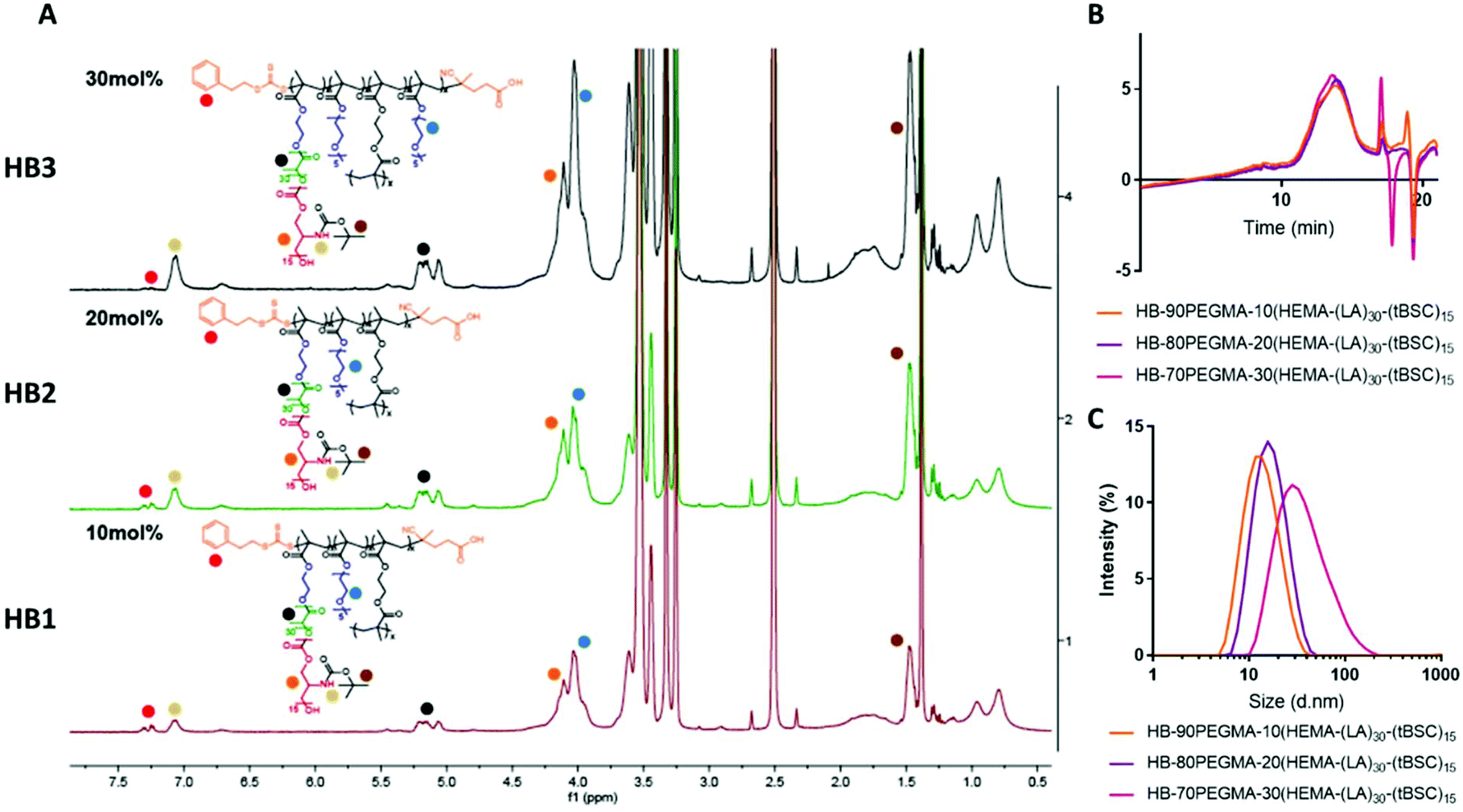 | ||
| Fig. 3 (A) 1H NMR spectra of the hyperbranched random copolymers synthesised in presence of PEGMA by RAFT polymerisation, (B) GPC traces of the polymers and (C) DLS traces of the unimolecular hyperbranched nanoparticles. | ||
Nanoparticle formation and dye encapsulation
Self-assembling amphiphilic PEG-polylactide and PEG-polycarbonate materials have been widely investigated as nanoparticle carriers in drug and gene delivery assays.42,43 Therefore, we screened the ability of the varying architecture polymers synthesised above to self-assemble into well-defined nanoparticles using the standard formulation technique of nanoprecipitation (solvent displacement).44 Rapid precipitation of the block polymers from acetone solutions into MilliQ water resulted in formation of nanoparticles with sizes varying from 70–144 nm (Table 2).| Sizea (nm) (at 500 μg ml−1) | Z-potentialb (mV) | Critical aggregation concentration (CAC) (μg ml−1) | |
|---|---|---|---|
| a Average values from at least 3 sample replicates produced from 3 batches. b Measurements in milliQ water. | |||
| P1. mPEG2000-(LA)50-(tBSC)50 | 115.8 ± 7.5 | −26.9 ± 2.5 | 9.5 |
| P2. mPEG5000-(LA)50-(tBSC)50 | 78.5 ± 3.5 | −23.5 ± 4.5 | 8.5 |
| P3. PEG4000-(LA)50-(tBSC)50 | 92.5 ± 4.5 | −10.7 ± 3.5 | 12.7 |
| P4. 3 armPEG1014-(LA)50-(tBSC)50 | 138.0 ± 8.0 | −29.2 ± 5.5 | 11.5 |
| P5. 4 armPEG797-(LA)50-(tBSC)50 | 123.5 ± 1.5 | −30.1 ± 5.5 | 12.1 |
To probe further the structures of the nanoparticles, representative samples were also imaged by TEM. The size ranges observed in the images were similar to those obtained by DLS, taking into account the normal compaction following the TEM drying process (Fig. S9†). Although the chain architectures were slightly different across the polymer series from P1 to P5, the morphologies of all the self-assembled systems were approximately spherical, with no evidence of worm-like micelles or other altered geometry superstructures. Further detail in structure was not observable from the micrographs owing to the limits in contrast obtained from non-stained samples and the resolution of the images. However, based on the ratios of hydrophobic to hydrophilic blocks, micelle-like nanoparticles would be expected to form under the conditions we employed, with vesicles or ‘worms’ only likely at higher polylactide and polycarbonate mole fractions owing to packing parameter constraints. The zeta potentials of all these nanoparticles were negative, below −10 mV, as expected based on the presence of a hydrophilic PEG corona shell and the weak lone-pair cation interactions at the oxyethylene units and the corresponding association of anions to form the outer layers (Table 2). The length of the hydrophobic fragments was kept constant across the range of copolymers, and thus the differences in size and zeta potential can be attributed to the different PEGylated initiators adopted. It was apparent that the A–B polymers based on PEG-lactide resulted in nanoparticles of a smaller size compared to the random copolymers, while the chain extended A–B–C block copolymer formed larger particles, perhaps due to difficulties in packing with the bulkier BOC-carbonate fragments (Fig. S10†). Critical Aggregation Concentration (CAC) values were calculated by exploiting the light scattering count rate intensity variation according to serial dilution experiments. The resulting values were in the range of 8.5 to 12.7 μg ml−1 which indicated that NPs were present at low concentration in the final suspensions and required high dilution before disaggregation occurred (Fig. S11†).
Well-defined nanoparticles have also been formed using branched polymers encapsulated within A–B linear copolymers, and their self-assembly and colloidal properties have been extensively studied previously.45,46 We therefore selected polymers P2 and P5 as the model PEGylated copolymers (one linear and one branched) to encapsulate HB2 through a co-nanoprecipitation step. DLS analysis of the individual NPs/HBs in aqueous solution showed a single population, and the control mixed solution of P2 + HB2 and P5 + HB2 showed two populations corresponding to the individual components as expected. Importantly, the co-nanoprecipitation systems showed only a single DLS peak, confirming that the HBs were able to be entrapped within the larger NPs (Fig. 4).
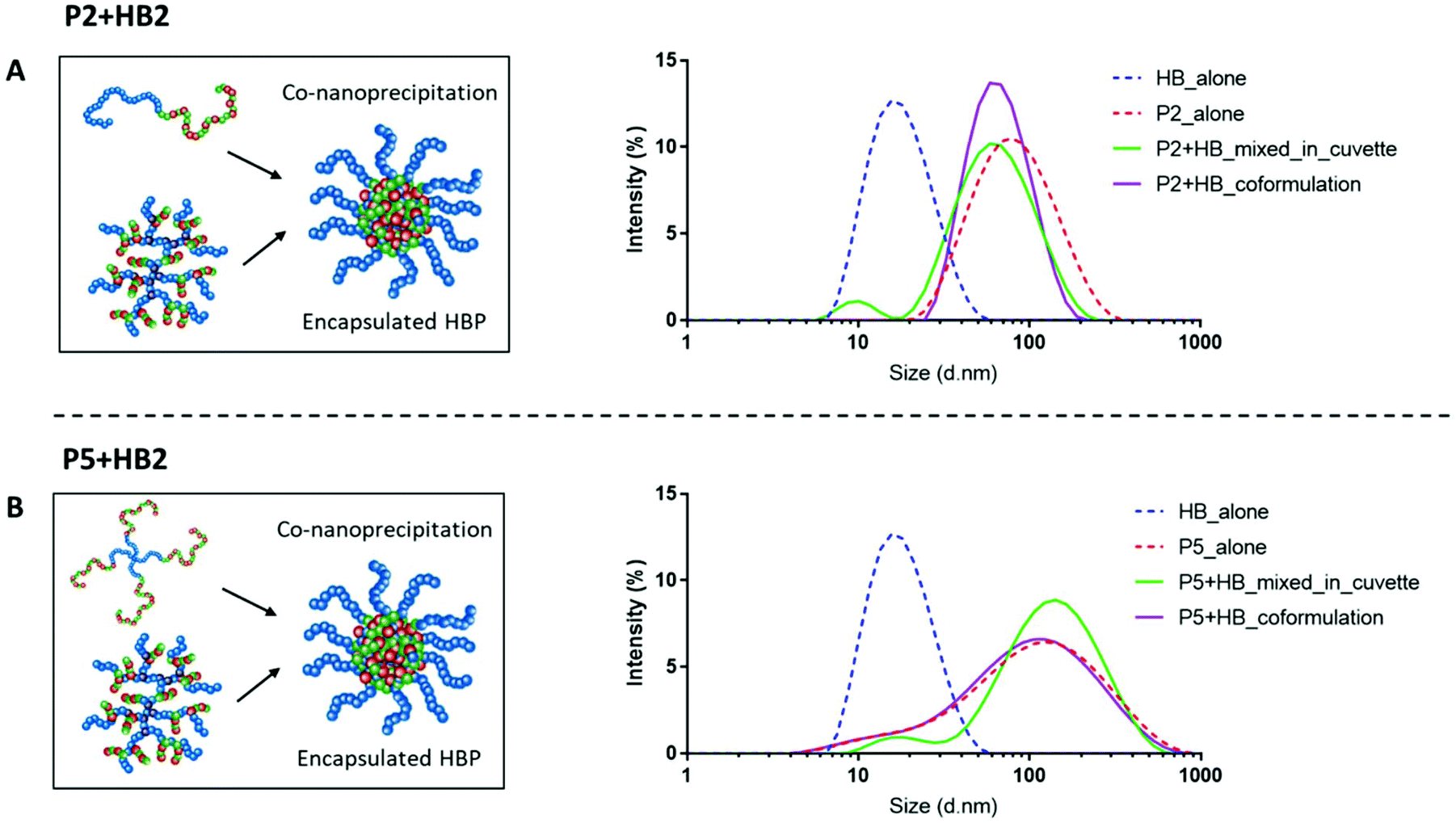 | ||
| Fig. 4 Co-nanoprecipitation of (A) P2 with HB2 and (B) P5 with HB2. (Left) schematics of co-nanoprecipitation process and (right) DLS traces confirming successful encapsulation of the HB. Dotted lines represent polymers alone, green traces represent the mixed in cuvette control and purple traces represent the successful coformulation. | ||
A second indicator of self-assembly, and a guide to utility in biomedical delivery applications, was the ability of the nanoparticles to encapsulate a probe or drug molecules. The polymer nanoparticles were thus screened for encapsulation of the dye 4-aminofluorescein in a preliminary assay. For these encapsulation experiments we selected the linear co-polymers and the star polymers P1–P5, in order to evaluate in the first instance the compatibilities of the PEG-poly(lactide)-poly(carbonate) ‘parent’ structures with a model drug. In Fig. S12† it can be seen that the star branched PEGylated copolymers, namely P4 and P5, encapsulated more of the dye than the other polymers, in agreement with data reported previously with similar materials.47–49
Polymer side-chain derivatisation and drug coupling
The intended use of the polymer family as delivery agents was also envisaged in which drugs were covalently attached as well as or in place of encapsulated therapeutics. Accordingly, polymer P2 was again chosen as the exemplar material, from the small library and activated for drug attachment by removal of the BOC group. It was important for this step to occur without causing degradation of the polyester and polycarbonate backbone, thus the reaction was performed at 0 °C. In experiments to evaluate the optimal timing of the reactions, it was found that consistent deprotection of up to 30% of the initial BOC functionality was reached within 30 minutes of reaction, without degradation of the polymer backbone as shown by NMR (Fig. 5). It was also found that removal of the BOC group and formation of free amine groups on the side-chains reduced the solubility of the polymer in CDCl3, thus all the deprotected materials were analysed in DMSO-d6 (for clarity also the unfunctionalized polymer was analysed in both the deuterated solvents as shown in Fig. S13†). As depicted in Fig. 3, the NH of the carbamate and the protons of the new produced ammonium moieties were clearly visible in the spectra between 7.0 and 8.6 ppm thus allowing simple calculation of the extent of BOC deprotection and conversion to free amine groups on the side chains.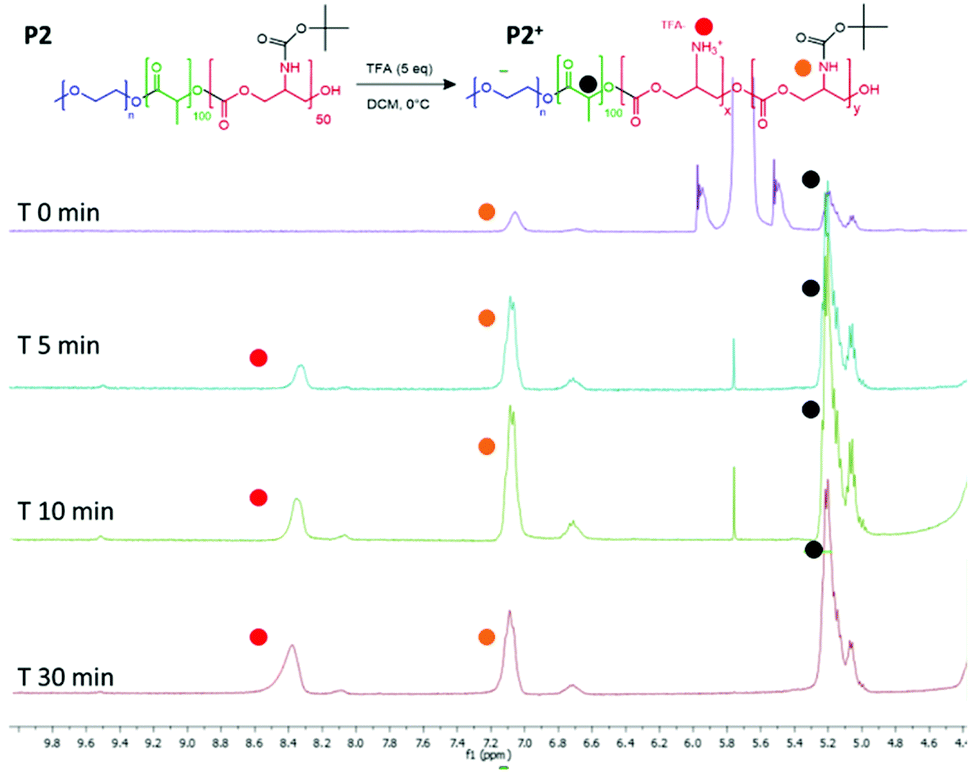 | ||
| Fig. 5 1H NMR spectra to show BOC deprotection on the carbonate unit in the polymer backbone. Spectra recorded after purification of the polymer from 0, 5, 10 and 30 minutes of reaction time. | ||
Following the formation of the amine groups on P2, coupling of the widely-used chemotherapeutic, doxorubicin was carried out using N,N′-disuccinimidyl-carbonate (DSC) as the coupling-agent, thus linking the drug to the polymer via a stable urea linkage. The resultant polymer prodrug P2-DOXO contained ∼10 molecules of doxorubicin based on NMR and UV analysis, indicating a coupling yield of ∼80% based on the 12–13 free NH2 units available per chain following BOC deprotection (Fig. 6). The final weight percentage of DOXO in the polymeric prodrug system varied by less than 10% when measured by the two spectroscopic techniques (e.g. for 10 units 18% w/w by NMR and 12% by UV-Vis). The polymer prodrug was subsequently formulated into nanoparticles, with a size range of 130–145 nm as indicated by DLS and confirmed by TEM (Fig. 6). The increase in size over the plain polymer nanoparticles was attributed to the change in chain packing due to the pendant doxorubicin molecules, while the zeta potential remained negative confirming an external PEG-corona (−19.0 ± 0.5 mV). Further indirect evidence of doxorubicin coupling was obtained from experiments in which the deprotected polymer (P2*) was formulated into nanoparticles prior to drug conjugation. As can be observed in Fig. S14,† two populations of nanoparticles were present, indicating disrupted self-assembly of the polymer due to the presence of the side-chain amines, and in contrast to the defined self-assembly of P2-DOXO. In addition, the zeta-potential of the P2* nanoparticle suspension was positive (+11 mV), again indicative of free amines and a likely inversion of the blocks of the polymer exposed in aqueous suspension.
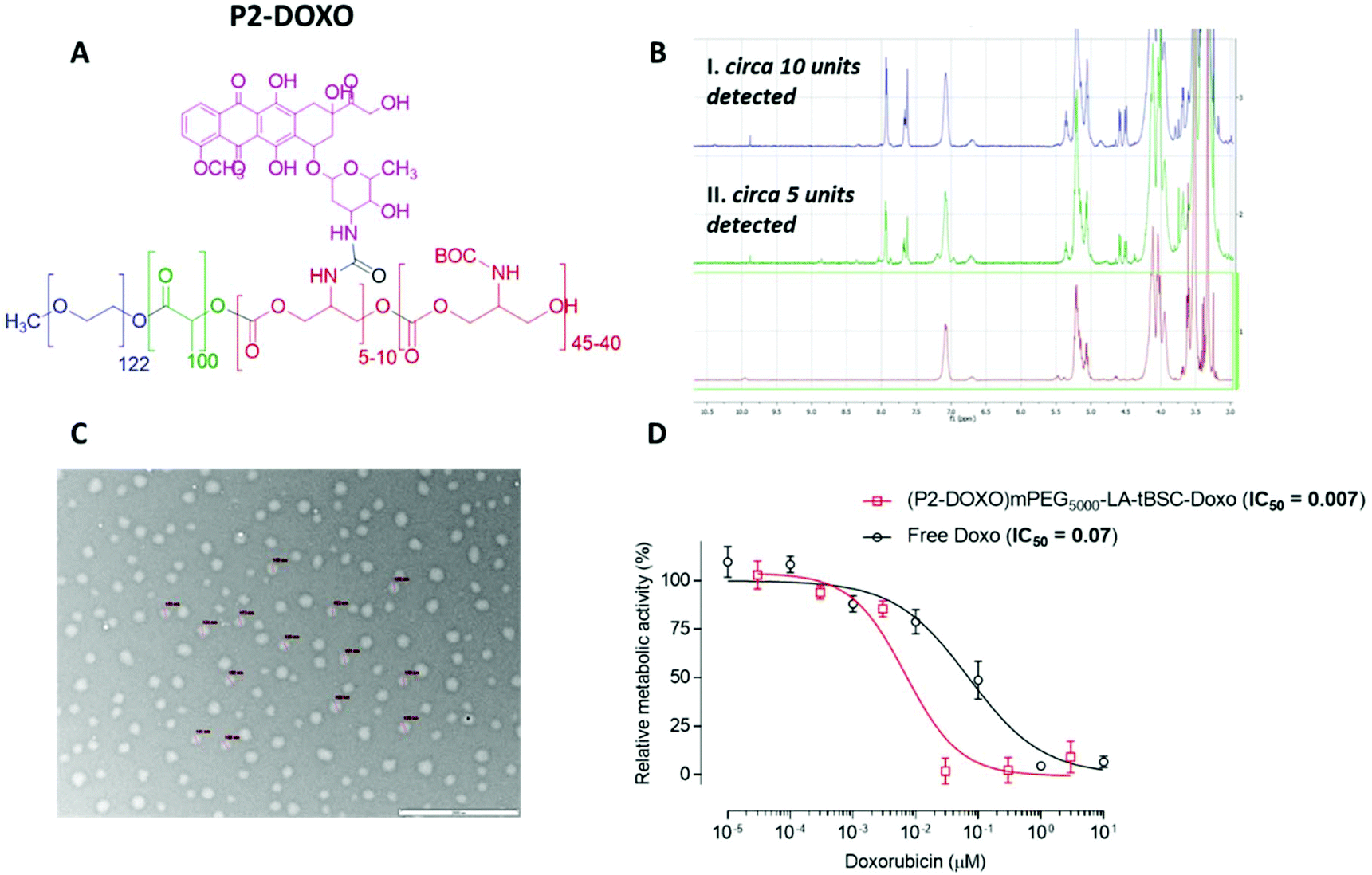 | ||
| Fig. 6 A. Structure of the polymeric prodrug mPEG5000-(LA)50-(tBSC)50-DOXO. B. Stacked 1HNMR spectra of the unmodified mPEG5000-(LA)50-(tBSC)50 (bottom) and two modifications of mPEG5000-(LA)50-(tBSC)50-DOXO (middle and top spectra) at two different extents of functionalisation. C. TEM micrographs of the mPEG5000-(LA)50-(tBSC)50-DOXO NPs prodrug. D. IC50 plots of the mPEG5000-(LA)50-(tBSC)50-DOXO against free doxorubicin in MCF7 cell line. | ||
Effects of polymer formulations on breast cancer cells
The intended uses of these materials in biomedical applications required a preliminary screen of their effects on measures of cellular viability. Polymer P2 was chosen as the control model nanoparticle formulation and was incubated with MCF7 breast cancer cells for up to 48 hours’ exposure. No change in cellular metabolic activity or plasma membrane integrity was observed after this time as evaluated by the PrestoBlue assay and monitoring extracellular release of LDH. These data indicated that the polymer ‘platform’ of PEG-poly(lactide)-co-poly(carbonate) was well-tolerated by the model breast cancer cells. In contrast, while the deprotected polymer, P2* demonstrated no adverse effects in MCF7 cells after 4 hours of exposure, concentration-dependent toxicity was elicited at exposures of 24 hours and longer (Fig. S15†). Cellular damage was characterised by plasma membrane disruption, as evidenced by release of LDH and a decline in metabolic activity, highlighting the lytic activity of the amphiphilic and positively charged copolymer formed after BOC-deprotection (Fig. S14†). We then evaluated the effects of the P2-DOXO polymer, using the conjugate bearing 10 molecules of doxorubicin per polymer chain. As expected, the polymer prodrug was highly cytotoxic, with an estimated IC50 of 0.007 ± 0.005 μM based on doxorubicin concentration. Intriguingly, these data indicated that the polymer pro-drug was significantly more toxic than the free drug (IC50 of doxorubicin ∼0.07 ± 0.01 μM) under the same assay conditions (Fig. 6).When considered together, the data in this study show a number of advantages for the polymers synthesised. Our aim was to explore facile routes to functionalised materials which are known to be biodegradable and which could be adaptable for a variety of therapeutic formats. The key to this concept was to establish if ring-opening polymerisations of cyclic esters and carbonates could take place under mild reaction conditions, in high yields and in short reaction times to generate the ‘platform’ polymers which might further serve as drug delivery agents. In essence, our aim was to combine and refine some existing chemistries rather than develop entirely new polymerisation or derivatisation methodologies. Thus, for the formation of PEGylated copolymers, which have been extensively studied as self-assembling NPs for drug delivery, it was necessary to develop a versatile and controllable synthesis with as few steps as possible, and minimal purification. We found that the use of DBU catalysed ROP enabled synthesis to take place within 30 minutes while retaining all the key functionality. In contrast, triazabicyclodecene (TBD) catalysed polymerisation was unsuitable, because although lactide ring-opening polymerisations occur rapidly with this catalyst (reaction time <60 s) there was also a tendency of TBD to cleave BOC-protected functionalities, leading to a fast self-immolative degradation of the growing polymer chain.19,50 Under the conditions we developed, DBU catalysed the polymer-forming reactions efficiently without any undesired side reactions. The utility of DBU was further exemplified by the use of HEMA as an initiator, resulting in a methacrylate-terminated poly(lactide)-co-poly(carbonate), and it is important to note that again the reaction times and conditions were critical in order to avoid transesterification reactions.51 The retention of the methacrylate group was demonstrated by a facile thiol–ene click reaction in high yield, but also in the use of the compound as a macromonomer in hyperbranched polymer synthesis. A number of papers have shown the advantages of hyperbranched polycarbonates in drug delivery,52–54 and thus we aimed to show that well-defined hyperbranched polymers could be produced in a single step from the macromonomer with minimal purification. This was easily achieved by RAFT polymerisation and three single-chain nanoparticle polymers were generated by this route, with dimensions of 12–30 nm. These polymers, and also the amphiphilic copolymers produced from PEG initiators were then rapidly assembled into kinetically trapped micellar-like nanoparticles by the ‘nanoprecipitation’ technique. Across the range of polymers, nanoparticles with hydrophilic outer ‘shells’ and varying from 12–140 nm in diameter were produced, and thus with properties appropriate for avoiding renal clearance and RES uptake, and for prolonging circulation times following systemic injection.
The culminating experiments evaluated some exemplar polymers for drug conjugation and effects on a model cell line. An important criterion for drug delivery is the loading of therapeutic molecules on a carrier, and for the linear PEG-lactide-carbonate polymer P2 after BOC deprotection and tagging to the formed amine side-chains, a molar ratio of 10–12 doxorubicin moieties per polymer chain was achieved. This polymer was found to be 10-fold more potent against MCF-7 cells in culture than the free drug, while the parent polymer before BOC deprotection was well-tolerated under all conditions tested. These results are a good indicator that the materials described in this paper may form a functionalisable polymer platform for subsequent in vivo evaluation.
Conclusions
In summation, we have shown data for these potential biomedical polymers which suggest a number of advantages compared to existing methodologies for biomaterial synthesis. Firstly, the use of DBU as catalyst for controlled ring-opening polymerisation enabled a range of polymers to be formed with good retention of functionality, and these materials were easily accessible through reactions under ambient and readily-achievable conditions. Secondly, both PEGylated and methacrylated hybrid copolymers could be generated with minimal purification steps, preserving the key functionalities along the polymeric backbone and at the terminal positions, allowing for further modifications with drug molecules and labels for desired pharmaceutical and medical applications. Finally, the polymers could be designed for particular architectures or self-assembly properties but without requiring multiple reactions steps, and their association into drug delivery nanoparticles was easily demonstrated with model drugs and a cancer-relevant cell line. Future work will explore the detailed in vitro and in vivo applications of selected polymers from this initial study.Data access statement
All raw data created during this research are openly available from the corresponding authors (vincenzo.taresco@nottingham.ac.uk, cameron.alexander@nottingham.ac.uk) and at the University of Nottingham Research Data Management Repository (https://rdmc.nottingham.ac.uk/) and all analysed data supporting this study are provided in the ESI† accompanying this paper.Funding sources
This work was supported by the Engineering and Physical Sciences Research Council [Grant Numbers EP/N006615/1, EP/N03371X/1, EP/L01646X/1] and the Royal Society [Grant WM150086] under a Wolfson Research Merit Award to CA.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors thank the Nanoscale and Microscale Research Centre (nmRC) for providing access to instrumentation. We also thank Esme Ireson, Paul Cooling and Kady Marriott for expert technical support.Notes and references
- R. Duncan, J. Drug Targeting, 2017, 25, 759–780 CrossRef CAS.
- R. Ferrari, M. Sponchioni, M. Morbidelli and D. Moscatelli, Nanoscale, 2018, 10, 22701–22719 RSC.
- J. H. Ko and H. D. Maynard, Chem. Soc. Rev., 2018, 47, 8998–9014 RSC.
- R. Duncan, J. Controlled Release, 2014, 190, 371–380 CrossRef CAS.
- S. Ahmed, S. B. Vepuri, R. S. Kalhapure and T. Govender, Biomater. Sci., 2016, 4, 1032–1050 RSC.
- G. Shim, M. G. Kim, D. Kim, J. Y. Park and Y. K. Oh, Adv. Drug Delivery Rev., 2017, 115, 57–81 CrossRef CAS.
- H. J. Wagner, A. Sprenger, B. Rebmann and W. Weber, Adv. Drug Delivery Rev., 2016, 105, 77–95 CrossRef CAS.
- X. Fan, Z. Li and X. J. Loh, Polym. Chem., 2016, 7, 5898–5919 RSC.
- R. Duncan and R. Gaspar, Mol. Pharm., 2011, 8, 2101–2141 CrossRef CAS.
- R. Gaspar and R. Duncan, Adv. Drug Delivery Rev., 2009, 61, 1220–1231 CrossRef CAS.
- V. Arias, P. Olsén, K. Odelius, A. Höglund and A. C. Albertsson, Polym. Chem., 2015, 6, 3271–3282 RSC.
- C. Jérôme and P. Lecomte, Adv. Drug Delivery Rev., 2008, 60, 1056–1076 CrossRef.
- L. A. Dailey, M. Wittmar and T. Kissel, J. Controlled Release, 2005, 101, 137–149 CrossRef CAS.
- K. Yu, Y. Zhou, Y. Li, X. Sun, F. Sun, X. Wang, H. Mu, J. Li, X. Liu, L. Teng and Y. Li, Biomater. Sci., 2016, 4, 1219–1232 RSC.
- Q. Yin, R. Tong, L. Yin, T. M. Fan and J. Cheng, Polym. Chem., 2014, 5, 1581–1585 RSC.
- J. Kasperczyk, S. Li, J. Jaworska, P. Dobrzyński and M. Vert, Polym. Degrad. Stab., 2008, 93, 990–999 CrossRef CAS.
- F. Danhier, E. Ansorena, J. M. Silva, R. Coco, A. Le Breton and V. Préat, J. Controlled Release, 2012, 161, 505–522 CrossRef CAS.
- M. E. Werner, N. D. Cummings, M. Sethi, E. C. Wang, R. Sukumar, D. T. Moore and A. Z. Wang, Int. J. Radiat. Oncol., Biol., Phys., 2013, 86, 463–468 CrossRef CAS.
- R. C. Pratt, B. G. G. Lohmeijer, D. A. Long, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2006, 128, 4556–4557 CrossRef CAS.
- W. W. Gerhardt, D. E. Noga, K. I. Hardcastle, A. J. García, D. M. Collard and M. Weck, Biomacromolecules, 2006, 7, 1735–1742 CrossRef CAS.
- A. Alla, J. Oxelbark, A. Rodríguez-Galán and S. Muñoz-Guerra, Polymer, 2005, 46, 2854–2861 CrossRef CAS.
- J. A. Wilson, S. A. Hopkins, P. M. Wright and A. P. Dove, Biomacromolecules, 2015, 16, 3191–3200 CrossRef CAS.
- S. Tempelaar, L. Mespouille, O. Coulembier, P. Dubois and A. P. Dove, Chem. Soc. Rev., 2013, 42, 1312–1336 RSC.
- H. Qian, A. R. Wohl, J. T. Crow, C. W. MacOsko and T. R. Hoye, Macromolecules, 2011, 44, 7132–7140 CrossRef CAS.
- B. G. G. Lohmeijer, R. C. Pratt, F. Leibfarth, J. W. Logan, D. A. Long, A. P. Dove, F. Nederberg, J. Choi, C. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 8574–8583 CrossRef CAS.
- W. Chin, G. Zhong, Q. Pu, C. Yang, W. Lou, P. F. De Sessions, B. Periaswamy, A. Lee, Z. C. Liang, X. Ding, S. Gao, C. W. Chu, S. Bianco, C. Bao, Y. W. Tong, W. Fan, M. Wu, J. L. Hedrick and Y. Y. Yang, Nat. Commun., 2018, 9, 917 CrossRef.
- A. Baroni, L. Vlaminck, L. Mespouille, F. Du Prez, N. Delbosc and B. Blankert, Macromol. Rapid Commun., 2019, 40, 1800743 CrossRef.
- X. Guo, B. Choi, A. Feng and S. H. Thang, Macromol. Rapid Commun., 2018, 39, 1800479 CrossRef.
- S. Venkataraman, N. Veronica, Z. X. Voo, J. L. Hedrick and Y. Y. Yang, Polym. Chem., 2013, 4, 2945–2948 RSC.
- F. Y. Qiu, C. C. Song, M. Zhang, F. S. Du and Z. C. Li, ACS Macro Lett., 2015, 4, 1220–1224 CrossRef CAS.
- W. Cheng, R. Rajendran, W. Ren, L. Gu, Y. Zhang, K. H. Chuang and Y. Liu, J. Mater. Chem. B, 2014, 2, 5295–5301 RSC.
- F. Y. Qiu, C. C. Song, M. Zhang, F. S. Du and Z. C. Li, ACS Macro Lett., 2015, 4, 1220–1224 CrossRef CAS.
- M. Biondi, D. Guarnieri, H. Yu, V. Belli and P. A. Netti, Nanotechnology, 2013, 24, 045101 CrossRef.
- B. Luan, T. Huynh and R. Zhou, J. Phys. Chem. Lett., 2015, 6, 331–337 CrossRef CAS.
- F. Luo, X. H. Meng, J. Fan, S. Z. Fu, J. M. Fan, J. B. Wu, K. Shi, Z. Y. Qian, Y. Qu, L. L. Yang and Z. Li, J. Biomed. Nanotechnol., 2013, 10, 427–435 Search PubMed.
- D. Andrade, C. Moya, F. Olate, N. Gatica, S. Sanchez, E. Díaz, E. Elgueta, M. Parra and M. Dahrouch, RSC Adv., 2016, 6, 38505–38514 RSC.
- L. Sun, A. Pitto-Barry, A. W. Thomas, M. Inam, K. Doncom, A. P. Dove and R. K. O'Reilly, Polym. Chem., 2016, 7, 2337–2341 RSC.
- N. Francini, L. Purdie, C. Alexander, G. Mantovani and S. G. Spain, Macromolecules, 2015, 48, 2857–2863 CrossRef CAS.
- L. Simón-Gracia, D. Pulido, C. Sevrin, C. Grandfils, F. Albericio and M. Royo, Org. Biomol. Chem., 2013, 11, 4109–4121 RSC.
- J. Zong, S. L. Cobb and N. R. Cameron, Biomater. Sci., 2017, 5, 872–886 RSC.
- M. Perfézou, A. Turner and A. Merkoçi, Chem. Soc. Rev., 2012, 41, 2606–2622 RSC.
- J. M. Rabanel, J. Faivre, S. F. Tehrani, A. Lalloz, P. Hildgen and X. Banquy, ACS Appl. Mater. Interfaces, 2015, 7, 10374–10385 CrossRef CAS.
- C. Garofalo, G. Capuano, R. Sottile, R. Tallerico, R. Adami, E. Reverchon, E. Carbone, L. Izzo and D. Pappalardo, Biomacromolecules, 2014, 15, 403–415 CrossRef CAS.
- D. Kakde, V. Taresco, K. K. Bansal, E. P. Magennis, S. M. Howdle, G. Mantovani, D. J. Irvine and C. Alexander, J. Mater. Chem. B, 2016, 4, 7119–7129 RSC.
- F. L. Hatton, L. M. Tatham, L. R. Tidbury, P. Chambon, T. He, A. Owen and S. P. Rannard, Chem. Sci., 2015, 6, 326–334 RSC.
- F. Y. Hern, A. Hill, A. Owen and S. P. Rannard, Polym. Chem., 2018, 9, 1767–1771 RSC.
- E. Lallana, R. Donno, D. Magrì, K. Barker, Z. Nazir, K. Treacher, M. J. Lawrence, M. Ashford and N. Tirelli, Int. J. Pharm., 2018, 548, 530–539 CrossRef CAS.
- D. J. A. Cameron and M. P. Shaver, Chem. Soc. Rev., 2011, 40, 1761–1776 RSC.
- J. Burke, R. Donno, R. D'Arcy, S. Cartmell and N. Tirelli, Biomacromolecules, 2017, 18, 728–739 CrossRef CAS.
- C. P. Easterling, T. Kubo, Z. M. Orr, G. E. Fanucci and B. S. Sumerlin, Chem. Sci., 2017, 8, 7705–7709 RSC.
- L. A. Ruiz-Cantu, A. K. Pearce, L. Burroughs, T. M. Bennett, C. E. Vasey, R. Wildman, D. J. Irvine, C. Alexander and V. Taresco, Macromol. Chem. Phys., 2019, 220, 1800459 CrossRef.
- H. zhen Jia, J. yi Zhu, X. li Wang, H. Cheng, G. Chen, Y. fang Zhao, X. Zeng, J. Feng, X. zheng Zhang and R. xi Zhuo, Biomaterials, 2014, 35, 5240–5249 CrossRef.
- H. Z. Jia, H. F. Wang, C. W. Liu, C. Li, J. Yang, X. D. Xu, J. Feng, X. Z. Zhang and R. X. Zhuo, Soft Matter, 2012, 8, 6906–6912 RSC.
- J. Sun, K. I. Aly and D. Kuckling, RSC Adv., 2017, 7, 12550–12560 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9bm00667b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |