
Modulating bioactivities of primary ammonium-tagged antimicrobial aliphatic polycarbonates by varying length, sequence and hydrophobic side chain structure†

Abstract
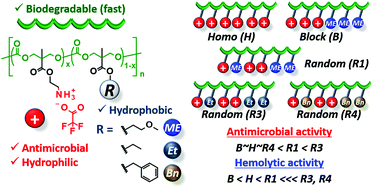
Cationic aliphatic polycarbonates bearing primary ammonium side chains have been developed with relatively high molecular weights and controlled macromolecular architectures. These polycarbonates exhibit reasonable antimicrobial activity against Gram-negative and Gram-positive bacteria. The prepared homopolymers could be effective against Gram-negative bacteria whose growth is usually inhibited by copolymers with hydrophobic comonomer units when quaternary ammonium salts (QAS) are used at the cationic side chains. A methoxyethyl (ME) side chain was explored as a comonomer unit for modulating biological activities, besides conventional hydrophobic side chains including ethyl and benzyl groups. In contrast to the ethyl side chain that increases both antimicrobial and hemolytic activities, the ME side chain serves to enhance the antimicrobial activity, but suppresses the hemolytic activity. This could be attributed to the unique characteristics of an aliphatic polycarbonate bearing a ME side chain: hemocompatibility, cell adhesion property, and selective interactions with proteins. The benefits of blood compatibility of the cationic aliphatic polycarbonates with the use of the primary ammonium side chains have been reported for the first time. The polycarbonate main chain is subjected to hydrolysis, which reduces the inherent cytotoxicity of polycations. This hydrolytic property is specific to these primary ammonium-tagged polycarbonates and could be an advantage over previously reported QAS-tagged antimicrobial polycarbonates.
- This article is part of the themed collection: Biomaterials Science Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

