
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced drug internalization and therapeutic efficacy of PEGylated nanoparticles by one-step formulation with anti-mPEG bispecific antibody in intrinsic drug-resistant breast cancer†
Yi-An
Cheng‡
 a,
I-Ju
Chen‡
b,
Yu-Cheng
Su
c,
Kai-Wen
Cheng
b,
Yun-Chi
Lu
a,
Wen-Wei
Lin
abde,
Yuan-Chin
Hsieh
b,
Chien-Han
Kao
a,
Fang-Ming
Chen
f,
Steve R.
Roffler
*ag and
Tian-Lu
Cheng
*abeh
a,
I-Ju
Chen‡
b,
Yu-Cheng
Su
c,
Kai-Wen
Cheng
b,
Yun-Chi
Lu
a,
Wen-Wei
Lin
abde,
Yuan-Chin
Hsieh
b,
Chien-Han
Kao
a,
Fang-Ming
Chen
f,
Steve R.
Roffler
*ag and
Tian-Lu
Cheng
*abeh
aGraduate Institute of Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan
bDrug Development and Value Creation Research Center, Kaohsiung Medical University, Kaohsiung, Taiwan
cDepartment of Biological Science and Technology, National Chiao Tung University, Hsinchu, Taiwan
dDepartment of Laboratory Medicine, School of Medicine, College of Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan
eDepartment of Medical Research, Kaohsiung Medical University Hospital, Kaohsiung, Taiwan
fDepartment of Surgery, Faculty of Medicine, College of Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan
gInstitute of Biomedical Sciences, Academia Sinica, Taipei, Taiwan. E-mail: sroff@ibms.sinica.edu.tw; Tel: +886 2 26523079
hDepartment of Biomedical Science and Environmental Biology, Kaohsiung Medical University, Kaohsiung, Taiwan. E-mail: tlcheng@kmu.edu.tw; Tel: +886 7 3121101 2697
First published on 28th June 2019
Abstract
For those patients with HER2-overexpressing breast cancer, treatment with PEGylated liposomal doxorubicin (PLD) is inefficacious due to the intrinsic low sensitivity to doxorubicin. A very large increase in drug accumulation by active targeting may enhance the therapeutic efficacy of PLD. We established a humanized bispecific antibody (BsAb; mPEG × HER2) which has dual specificity for methoxy-polyethylene glycol (mPEG) and human epidermal growth factor receptor 2 (HER2) to enhance the specificity, internalization and anticancer activity of PLD for cancer cells that overexpress HER2. One-step formulation of PLD with mPEG × HER2 converted the PLD into HER2 targeted liposomes that were stable at 4 °C in PBS as well as at 37 °C in the presence of serum. αHER2/PLD induced receptor-mediated endocytosis and enhanced doxorubicin accumulation in MCF7/HER2 (HER2-amplified) breast cancer cells. αHER2/PLD also displayed more than 200-fold increased cytotoxicity to MCF7/HER2 cells and 28-fold increased cytotoxicity to drug-resistant MDA-MB-361 cells with a physical deletion of the TOP2A gene. αHER2/PLD specifically accumulated doxorubicin in the nucleus of cancer cells in tumor-bearing mice and produced significantly greater antitumor activity against MCF7/HER2 (P < 0.0001) and MDA-MB-361 (P < 0.05) tumors as compared to untargeted PLD. Furthermore, the cardiotoxicity of αHER2/PLD was similar to that of PLD in human cardiomyocytes and in mice. Our results indicate that the one-step formulation of PLD by mPEG × HER2 is a simple method to confer tumor specificity, increase drug internalization and enhance the anticancer activity of PLD against HER2-overexpressing and doxorubicin-resistant breast cancer.
Introduction
Coating the surface of nanoparticles with polyethylene glycol, known as “PEGylation”, is a common approach to reduce capture by the reticuloendothelial system1,2 and prolong the systemic circulation of nanomedicines.3,4 PEGylated nanoparticles (PEG-NPs) have been widely developed to encapsulate various therapeutic agents and imaging probes including small molecules, proteins and nucleic acids.3,5 Various PEG-NPs are under clinical development and two types have been approved including PEGylated liposomal doxorubicin (PLD; Caelyx® and Lipo-Dox®) for metastatic breast cancer, ovarian cancer and AIDS-related Kaposi's sarcoma, and PEGylated liposomal irinotecan (ONIVYDE®) for pancreatic cancer.6 PEG-NPs can passively accumulate in tumors via the enhanced permeability and retention (EPR) effect, thereby reducing off-target side effects.7 Thus, the overall risk of cardiotoxicity is significantly lower in patients treated with PLD as compared to patients treated with doxorubicin.8 However, although PLD can preferentially accumulate in tumors, PLD mainly remains in the extracellular stroma9 and slow release of doxorubicin in the extracellular matrix results in low cellular uptake.10 Reduced doxorubicin accumulation in tumor cells can reduce antitumor activity, especially for drug-resistant cancer cells, such as those that display intrinsic low sensitivity to doxorubicin due to deletion of DNA topoisomerase II alpha (TOP2A).11,12 Thus, developing an actively targeted form of PLD may improve drug internalization and anticancer efficacy against drug-resistant tumors.Chemical conjugation of antibodies, ligands and peptides to PLD can enhance specific targeting and uptake by tumor cells with concomitant improvement in therapeutic efficacy13–16 and imaging sensitivity.17–20 For breast cancer, HER2 (ERBB2) is emerging as a promising target for genomically informed therapy.21 Thus, for overcoming drug resistance, chemical conjugation of anti-HER2 antibodies on PLD increased nuclear doxorubicin accumulation and growth inhibition of BT-474/multidrug-resistant tumors.22 However, chemical conjugation of antibodies to PLD are limited by improper coupling orientation and antibody dysfunction. Conjugation of antibodies via random functional groups (amino, carboxyl, thiol groups) to PEG-lipids can result in heterogeneous coupling orientations,23,24 leading to loss in antibody binding activity.25 Site-specific conjugation can be achieved by addition of cysteine residues to the C-terminus of recombinant antibodies for coupling to maleimide groups introduced to the termini of PEG-lipids;26 however, free cysteines are susceptible to disulfide-mediated multimerization during production and storage unless reducing agents are used,27 which can in turn cause additional damage and loss of antibody stability.28 In addition, the high temperatures required for efficient post-insertion of scFv-PEG-lipids in liposomes can denature and reduce the activity of antibodies.13,29 Moreover, the steps of chemical modification are complicated. In order to prevent the reduction of stability and function of antibody and nanoparticle, non-covalent modification has been developed. Bifunctional adaptors, such as protein G, biotin and streptavidin, are conjugated on nanoparticles and then the adaptors can integrate antibodies on particles by a one-step formulation.30,31 Nevertheless, the adaptors also need to chemically couple on mPEG-probes in advance and the use of exogenous bifunctional adaptors is not allowed in the human body because of induction of immunogenicity. Developing antibody-conjugated nanomedicines with simple and homogeneous coupling orientations to increase cell uptake is important for the treatment of drug-resistant cancer.
To easily create nanomedicines that can target HER2-overexpressing cancer cells, we generated a humanized bispecific antibody (BsAb; mPEG × HER2) composed of a humanized anti-mPEG Fab linked to a human anti-HER2 single-chain antibody (scFv). This BsAb can bind to the terminal methoxy groups present on PEG chains surrounding PEGylated medicines to confer HER2-binding specificity to nanoparticles. Simply mixing mPEG × HER2 with PLD is expected to enhance targeting, endocytosis, and anti-cancer activity of PLD to HER2-overexpressing and TOP2A-deleted breast cancer cells (Fig. 1). We investigated the effect of varying the number of mPEG × HER2 molecules on PLD towards HER2 binding, drug uptake and nanomedicine stability. We then evaluated whether αHER2/PLD could increase endocytosis, doxorubicin uptake and cytotoxicity in MCF/HER2 cells. We further used TOP2A-deleted breast cancer cells, MDA-MB-361, to examine if targeted PLD was effective against doxorubicin-resistant cells. The tumor accumulation and therapeutic efficacy of αHER2/PLD in mice bearing MCF7/HER2 and MDA-MB-361 tumors were investigated. We also examined the cardiotoxicity αHER2/PLD in human cardiomyocytes and normal mice. We find that the one-step formulation of targeted PLD with mPEG × HER2 is a simple and stable method to confer HER2-specific targeting and enhanced therapeutic efficacy against doxorubicin-resistant breast cancer.
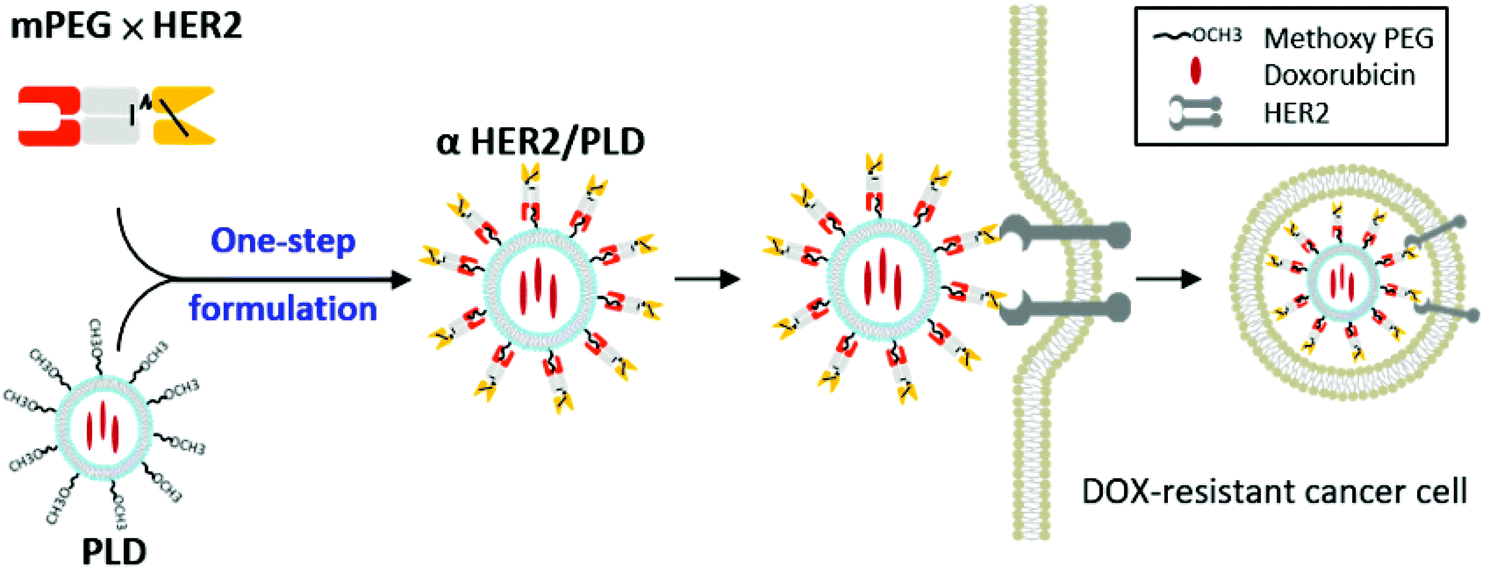 | ||
| Fig. 1 Strategy of anti-PEG bispecific molecules for targeted cancer therapy. The anti-PEG bispecific antibody (mPEG × HER2) can bind to the surface of PEGylated liposomal doxorubicin (PLD) to form αHER2/PLD, which confers HER2 specificity, and enhances internalization and anticancer activity against HER2-expressing and doxorubicin-resistant breast cancer cells. | ||
Results
Expression and function of humanized bispecific antibodies
To generate a BsAb with dual specificity for mPEG and HER2, we constructed mPEG × HER2 by fusing an anti-HER2 scFv to the Fab fragment of the humanized anti-mPEG antibody 15-2b, which can specifically target the methoxy-end of PEG.32 A negative control BsAb, mPEG × DNS, was created by exchanging the anti-HER2 scFv with an anti-DNS scFv, which binds the small chemical hapten dansyl (Fig. 2A). The humanized BsAbs were expressed in Expi-293 cells and purified on the mPEG750-coupling of CNBr-activated sepharose. The purity and molecular weight of mPEG × HER2 and mPEG × DNS were analyzed by SDS-PAGE. Fig. 2B shows that BsAbs were composed of a heavy-chain single-chain Fv fragment (H-scFv, 55 kDa) and a light-chain fragment (27 kDa) under reducing conditions, and a disulfide-linked BsAb (82 kDa) under non-reducing conditions. Analysis of mPEG × HER2 binding to mPEG by BLItz® system showed that the equilibrium dissociation constant was 8.667 × 10−8 M. To determine if mPEG × HER2 displays dual specificity to HER2 and mPEG, MCF7/HER2 cells were incubated with various concentrations of mPEG × HER2 or mPEG × DNS, then treated with PLD. Bound PLD was detected by sequential addition of the 6.3 anti-PEG backbone antibody, HRP-conjugated antibody and ABTS substrate, and the absorbance 405 nm was analyzed. Fig. 2C shows that mPEG × HER2 but not mPEG × DNS can simultaneously bind mPEG and HER2-overexpressing cells.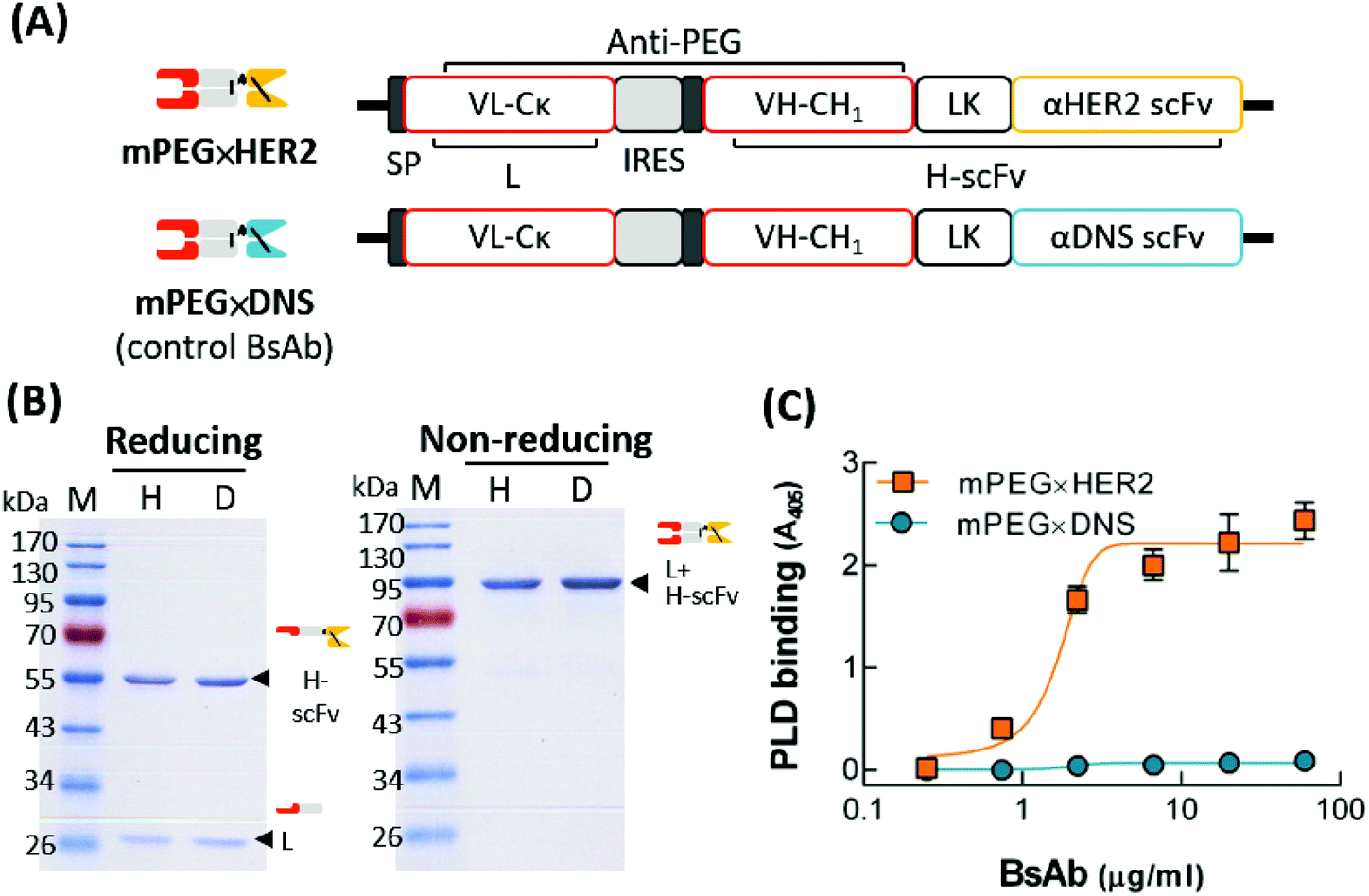 | ||
Fig. 2 Characterization of mPEG × HER2 and mPEG × DNS. (A) The gene constructs of BsAbs are composed of a signal peptide (SP), the anti-mPEG VL-Cκ (L), the anti-mPEG VH-CH1 (H), a flexible linker peptide (LK) and an anti-HER2 scFv (mPEG × HER2) or control anti-DNS scFv (mPEG × DNS). (B) The SDS-PAGE of purified mPEG × HER2 (H) or mPEG × DNS (D) under reducing (left) and non-reducing (right) conditions. (C) The mPEG and HER2 functions of mPEG × HER2 ( ) or mPEG × DNS ( ) or mPEG × DNS ( ) on MCF7/HER2 cells were detected via ELISA (n = 3). Bars, SD. ) on MCF7/HER2 cells were detected via ELISA (n = 3). Bars, SD. | ||
Optimal ratio of BsAbs for specific targeting of αHER2/PLD
To optimize the BsAb to mPEG modification ratio for PLD, mPEG × HER2 was mixed with PLD at BsAb densities on PLD of 13, 25, 50, 100 and 200 BsAb/PLD. We confirmed that the conjugation rate of each BsAb on PLD was more than 98% (Fig. S1†). We first compared the binding ability of the different BsAb-modified αHER2/PLDs to MCF7/HER2 cells. Fig. 3A shows that the absorbance value increased as the density of BsAb on PLD increased, indicating that αHER2/PLD displayed maximum binding at a BsAb/PLD ratio of 200 BsAb/PLD. We further investigated which αHER2/PLD produced the greatest uptake of doxorubicin into cancer cells. MCF7/HER2 cells were incubated with different ratios of αHER2/PLDs for 24 h before the cells were trypsinized to remove extracellular particles, and intracellular doxorubicin fluorescence intensity was then measured by flow cytometry. Fig. 3B shows that the fluorescence intensity of doxorubicin increased until a plateau was reached at a BsAb/PLD ratio of about 100 BsAb/PLD. This result suggests that there is no need to increase the BsAb density on PLD above 100 BsAb/PLD to achieve efficient drug uptake of αHER2/PLD into target HER2-overexpressing cells. Hence, we chose the BsAb density on PLD of 100 BsAb/PLD for the following studies.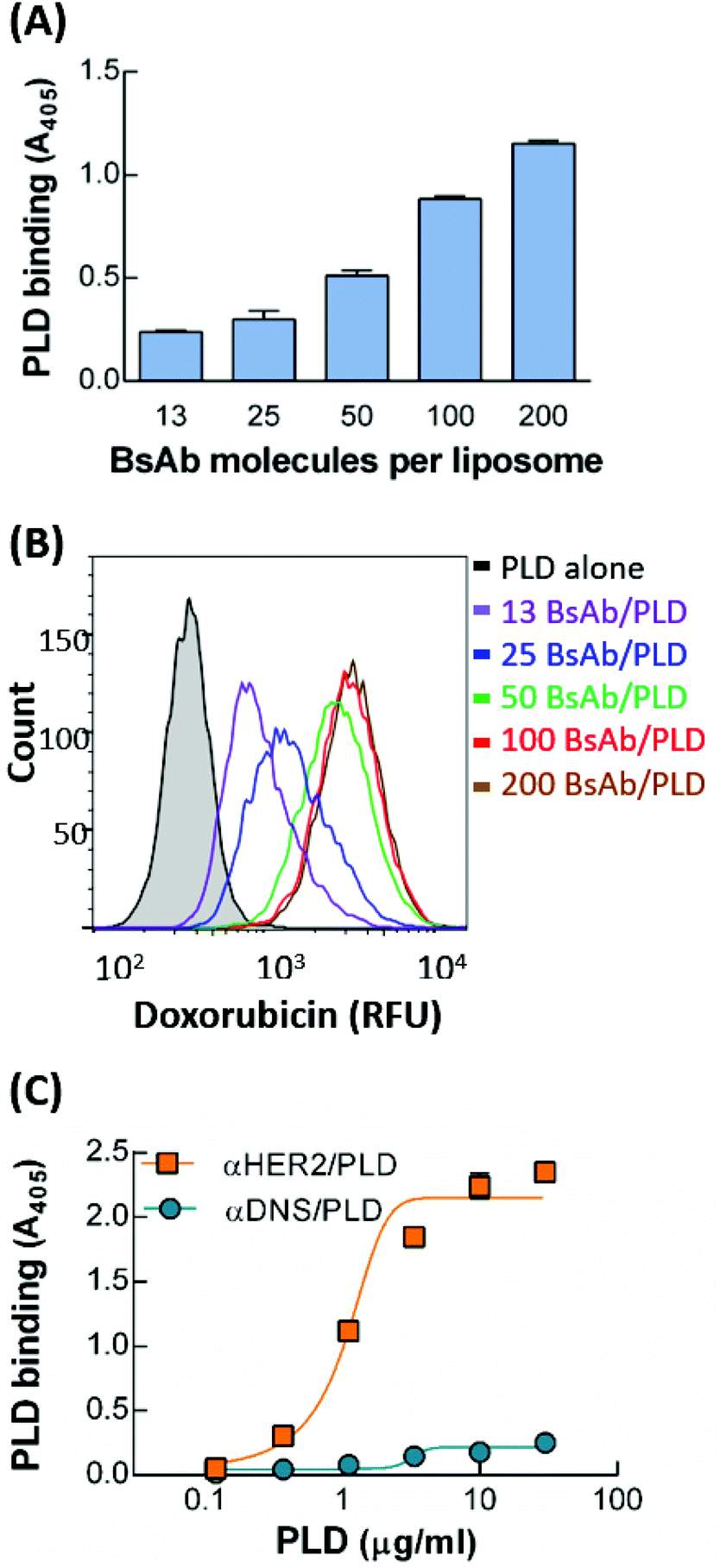 | ||
Fig. 3 Optimization of BsAb-modified PLD. (A) The HER2 targeting of αHER2/PLD with densities of BsAb on PLD of 13, 25, 50, 100 and 200 BsAb/PLD on MCF7/HER2 cells was investigated via ELISA (n = 3). Bars, SD. (B) The doxorubicin uptake of PLD or αHER2/PLD with different densities of BsAb on PLDs in MCF7/HER2 cells was measured from the doxorubicin fluorescence inside cells via flow cytometry. (C) The HER2 binding ability of αHER2/PLD ( ) or αDNS/PLD ( ) or αDNS/PLD ( ) at a BsAb/PLD density of 100 on MCF7/HER2 cells was determined via ELISA (n = 3). Bars, SD. ) at a BsAb/PLD density of 100 on MCF7/HER2 cells was determined via ELISA (n = 3). Bars, SD. | ||
The specificity of αHER2/PLD with the optimized BsAb ratio was examined by incubating MCF7/HER2 cells with various concentrations of αHER2/PLD or αDNS/PLD, then measuring the amount of surface-bound PLD by ELISA. Fig. 3C shows αHER2/PLD but not αDNS/PLD bound to HER2-overexpressing cells. The physical characteristics of mPEG × HER2 with 100 BsAb/PLD (αHER2/PLD) were described in previous studies, where the particle size of αHER2/PLD was slightly greater than that of PLD (105 nm versus 98.8 nm) and the zeta potential of αHER2/PLD was also slightly more negative than that of PLD (−50.8 mA versus −43.4 mA).33 This means that the formulation of αHER2/PLD slightly changed the particle size of PLD but the particle stability might not be influenced. To evaluate the stability of mPEG × HER2 on PLD, we examined the ability to bind MCF7/HER2 cells after storage at 4 °C for 7 days or incubation in 10% human serum for 72 h at 37 °C. The result also showed that αHER2/PLD retained the HER2 binding ability (Fig. S2A and S2B†). In addition, we found that the free BsAb of αHER2/PLD did not increase at 4 °C for 7 days (Fig. S2C†). We further followed the method34 of particle stability from covalent conjugation of particles to detect the particle size of αHER2/PLD in PBS at 4 °C for 7 days post-incubation with a Horiba SZ-100. The result showed that αHER2/PLD retained the particle size after 7 days incubation (Fig. S2D†). Thus, we demonstrated that αHER2/PLD is stable under storage and serum conditions.
Internalization and drug accumulation of αHER2/PLD via receptor-mediated endocytosis
The internalization of αHER2/PLD was examined by incubating αHER2/PLD, αDNS/PLD or PLD with MCF7/HER2 cells at 4 °C for 1 h before incubating each group at 4 °C or 37 °C for 3 h. PLD on the cell surface was detected by staining for the presence of PEG using flow cytometry. Fig. 4A shows that relatively high levels of αHER2/PLD were found on cells as compared with PLD and αDNS/PLD at 4 °C. αHER2/PLD levels on MCF7/HER2 cells gradually decreased with longer incubation times at 37 °C as compared at 4 °C, indicating that αHER2/PLD internalized into MCF7/HER2 cells. To determine if mPEG × HER2 induced receptor-mediated endocytosis of PEG-NPs in HER2-overexpressing cells, mPEG × HER2 and mPEG × DNS were mixed with PEGylated Qdot655 nm to form αHER2/Qdot655 nm and αDNS/Qdot655 nm, respectively. MCF7/HER2 cells were stained with LysoTracker Green and Hoechst 33342 to monitor lysosomes and nuclei, respectively. Cells were then incubated with αHER2/Qdot655 nm or αDNS/Qdot655 nm at 37 °C, and the fluorescence of Qdot655 nm (red), lysosomes (green) and nuclei (blue) was observed by real-time confocal microscopy to monitor binding, internalization and location in cells. Red fluorescence of αHER2/Qdot655 nm was detected on the cell surface at 60 min (Fig. 4B). After 130 min, yellow fluorescence was observed, indicating the co-localization of Qdot655 nm and lysosomes in MCF7/HER2 cells. In contrast, the red fluorescence was not observed with αDNS/Qdot655 nm. Next, the doxorubicin accumulation was examined by incubating αHER2/PLD with MCF7/HER2 cells at 37 °C as measured by intracellular fluorescence. αHER2/PLD enhanced the accumulation of doxorubicin in MCF7/HER2 cells whereas no obvious accumulation of doxorubicin was observed in cells treated with αDNS/PLD and PLD (Fig. 4C). These results show that αHER2/PLD displayed enhanced drug internalization and accumulation via receptor-mediated endocytosis in HER2-overexpressing cells.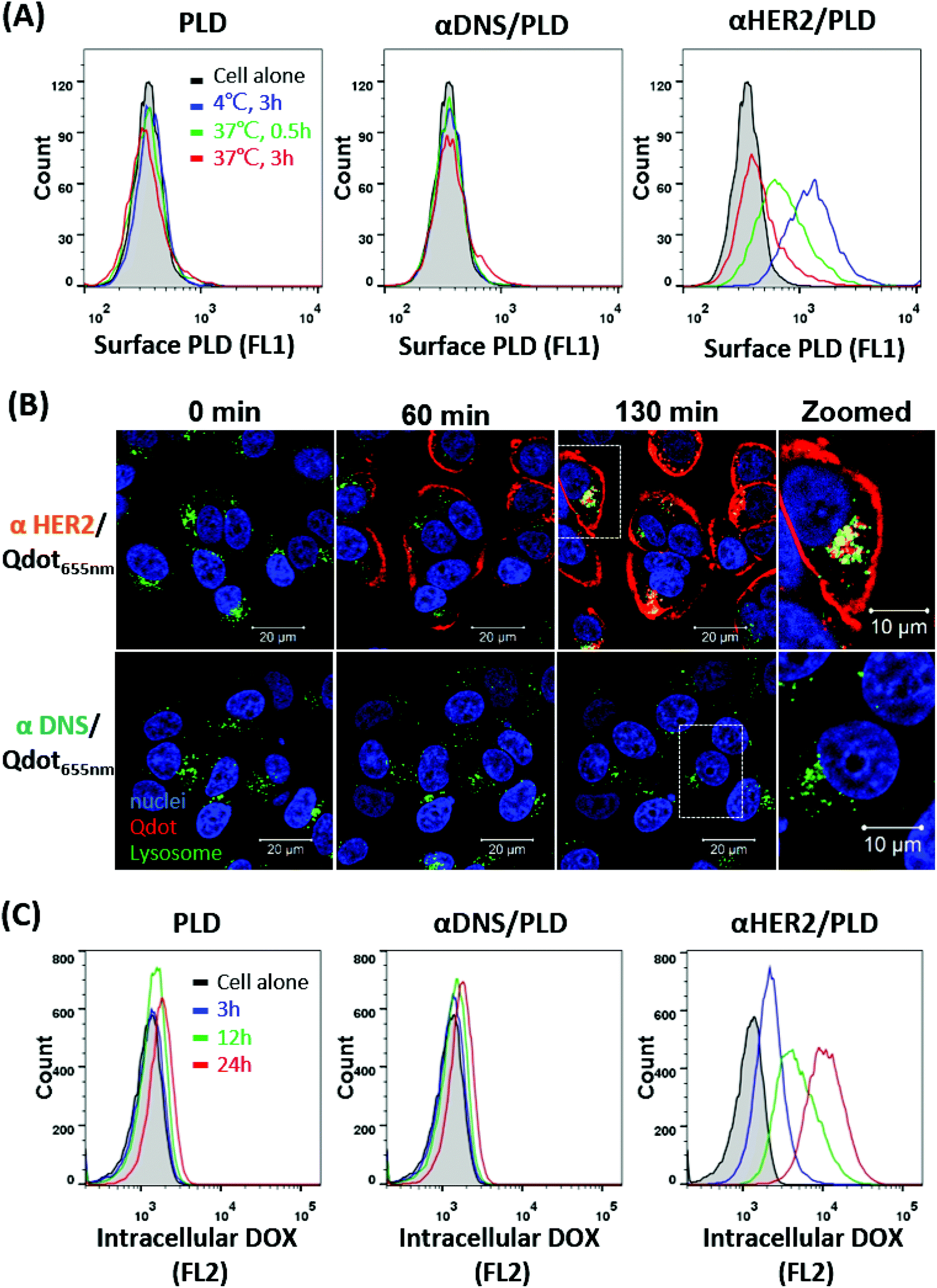 | ||
| Fig. 4 The internalization and drug uptake of αHER2/PLD in HER2-overexpressing cells. (A) The internalization of PLD (left), αDNS/PLD (middle) and αHER2/PLD (right) into MCF7/HER2 cells. The surface PLD was measured via flow cytometry using an anti-PEG antibody. (B) The co-localization of αHER2/Qdot655 nm or αDNS/Qdot655 nm (red) with lysosome (green) in MCF-7/HER2 cells was observed in real time by confocal microscopy. Merged images are shown. (C) The doxorubicin uptake of PLD (left), αDNS/PLD (middle) and αHER2/PLD (right) in MCF7/HER2 cells was detected via flow cytometry. | ||
αHER2/PLD displays enhanced cytotoxicity to HER2-overexpressing and doxorubicin-resistant breast cancer cells
The cytotoxicity of αHER2/PLD to three breast cancer cell lines was examined. MCF7/HER2 cells express high levels of HER2 and are sensitive to doxorubicin treatment, MCF7/neo1 cells express low levels of HER2 and are sensitive to doxorubicin treatment, and MDA-MB-361 cells express high levels of HER2 and are resistant to doxorubicin treatment (TOP2A gene deletion).11 These breast cancer cell lines were incubated with αHER2/PLD, αDNS/PLD or PLD for 3 h, the cells were washed and then the cell viability was determined by the ATPlite assay after 96 h. Fig. 5A shows that αHER2/PLD significantly reduced the viability of MCF7/HER2 cells as compared to PLD and αDNS/PLD with an IC50 value (0.07 μg mL−1) that was approximately 209-fold lower than that for PLD (IC50 = 14.6 μg mL−1) (Fig. 5D). As expected, the cytotoxicity of αHER2/PLD was similar to that of PLD and αDNS/PLD against MCF7/neo1 cells, which express very low levels of HER2 (Fig. 5B). Importantly, αHER2/PLD displayed enhanced cytotoxicity to doxorubicin-resistant MDA-MB-361 cells (IC50 = 1.89 μg mL−1) in comparison to PLD (IC50 = 52.2 μg mL−1) (Fig. 5C and D) Thus, these results demonstrate that αHER2/PLD increased the cytotoxicity of PLD in HER2-overexpressing cancer cells that were sensitive or resistant to doxorubicin.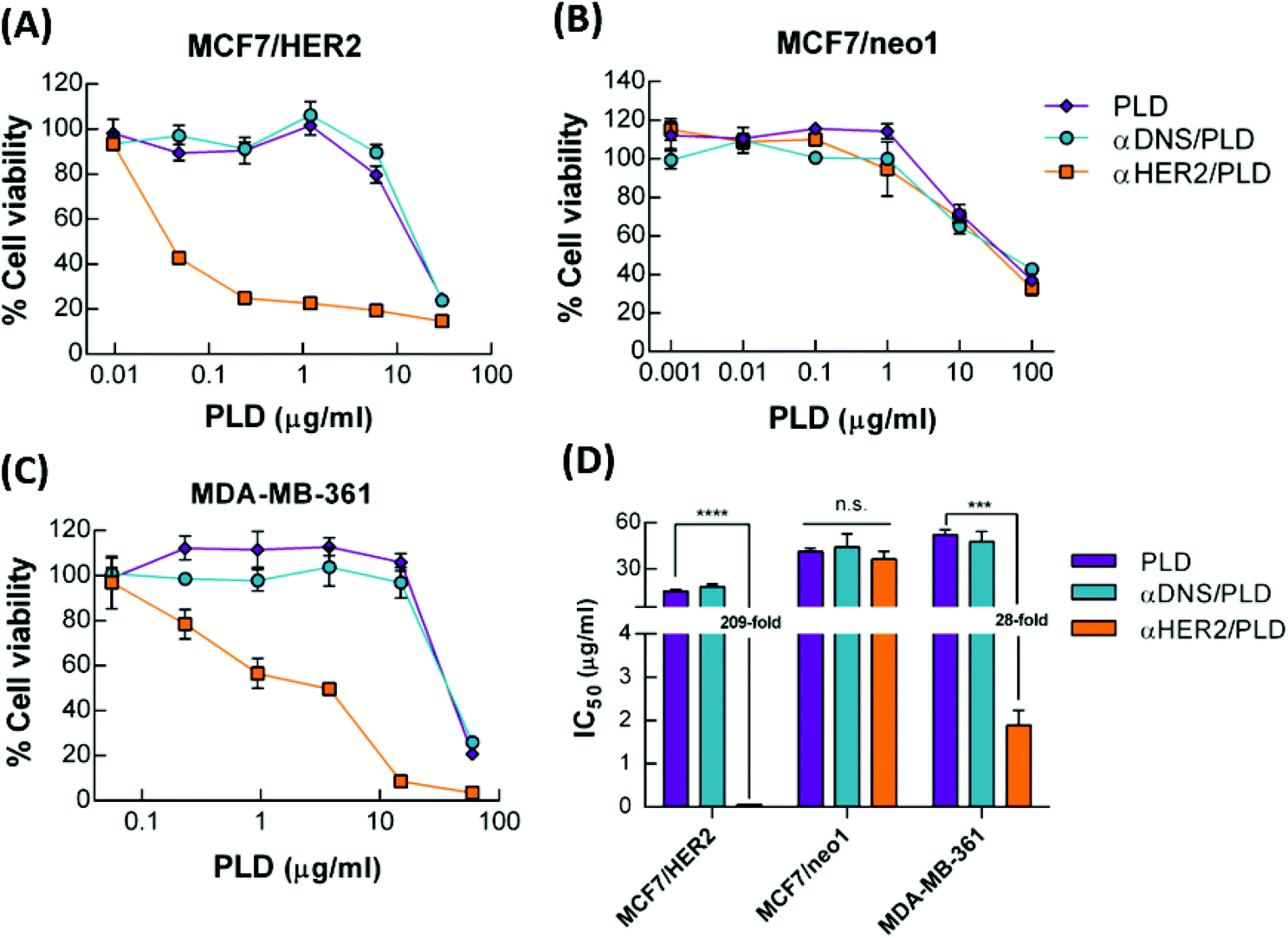 | ||
Fig. 5 The cytotoxicity of αHER2/PLD in HER2-overexpressing and doxorubicin-resistant breast cancer cells. PLD ( ), αDNS/PLD ( ), αDNS/PLD ( ) or αHER2/PLD ( ) or αHER2/PLD ( ) were incubated with (A) MCF7/HER2, (B) MCF7/neo1 and (C) MDA-MB-361 cancer cells for 3 h. The cell viability was determined by ATPlite analysis and the mean luminescence values compared to untreated control cells (n = 4). Bars, SD. (D) The half-maximal inhibitory concentration (IC50) values of MCF7/HER2, MCF7/neo1 and MDA-MB-361 cells treated with PLD, αDNS/PLD or αHER2/PLD. Bars, SD. ***P < 0.001, ****P < 0.0001; n.s., not significant (one-way ANOVA analysis). ) were incubated with (A) MCF7/HER2, (B) MCF7/neo1 and (C) MDA-MB-361 cancer cells for 3 h. The cell viability was determined by ATPlite analysis and the mean luminescence values compared to untreated control cells (n = 4). Bars, SD. (D) The half-maximal inhibitory concentration (IC50) values of MCF7/HER2, MCF7/neo1 and MDA-MB-361 cells treated with PLD, αDNS/PLD or αHER2/PLD. Bars, SD. ***P < 0.001, ****P < 0.0001; n.s., not significant (one-way ANOVA analysis). | ||
αHER2/PLD enhances delivery to HER2-overexpressing tumors
To investigate whether mPEG × HER2 can enhance tumor accumulation of PEGylated liposomal DiR (Lipo-DiR) in HER2-overexpressing tumors, we first mixed mPEG × HER2 and mPEG × DNS with far red-labeled liposomes (Lipo-DiR) to form αHER2/Lipo-DiR and αDNS/Lipo-DiR, respectively. Mice bearing MCF7/HER2 (HER2high, in left flank) and MCF7/neo1 (HER2low, in right flank) tumors were intravenously injected with αHER2/Lipo-DiR, αDNS/Lipo-DiR or Lipo-DiR, and then the fluorescence of DiR was detected by IVIS imaging at 24 h, 48 h and 72 h. The fluorescence signal of αHER2/Lipo-DiR was enhanced in MCF7/HER2 (HER2high) tumors as compared to MCF7/neo1 (HER2low) tumors from 24 h to 72 h after probe injection (Fig. 6A and B). We collected the tumors at 72 h, and we measured and calculated the relative region of interest (ROI) by total ROI of MCF7/HER2 divided by that of MCF7/neo1. The relative ROI of αHER2/Lipo-DiR was 3.0-fold (3.7 × 109versus 1.2 × 109) whereas αDNS/Lipo-DiR and Lipo-DiR produced 1.6-fold (3.6 × 109versus 2.3 × 109) and 1.8-fold (4.1 × 109versus 2.3 × 109), respectively (Fig. 6C). The results showed that αHER2/Lipo-DiR did not significantly enhance the ROI in MCF7/HER2 as compared to αDNS/Lipo-DiR and Lipo-DiR; however, it could enhance the relative ROI. These result suggested that all nanoparticles may be accumulated at a tumor site by the EPR effect, but αHER2/Lipo-DiR could increase the specific targeting on HER2-overexpressing tumors. We further demonstrated that the fluorescence intensity is not significantly different between αHER2/Lipo-DiR and Lipo-DiR in liver, heart, spleen and ovary whereas it is reduced for lung and kidney (Fig. S3†), indicating that αHER2/Lipo-DiR did not increase the off-target effect of PLD in mice.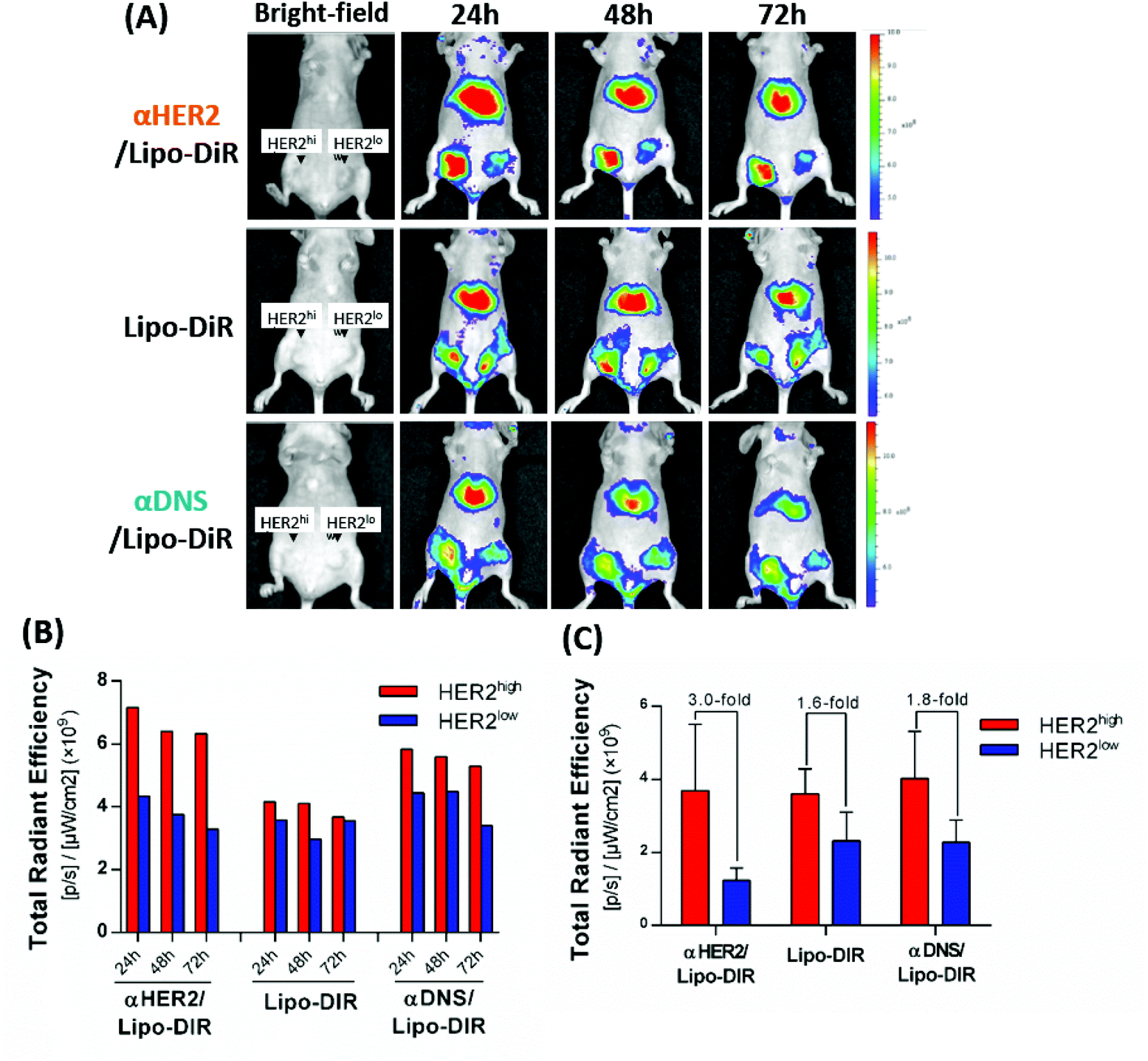 | ||
| Fig. 6 The tumor delivery of αHER2/PLD in HER2-overexpressing tumors. (A) αHER2/Lipo-DiR, Lipo-DiR or αDNS/Lipo-DiR were intravenously injected in mice bearing HER2high (left flank) and HER2low (right flank) tumors. The fluorescence intensity of DiR was detected at 24 h, 48 h, and 72 h after injection by IVIS. (B) Quantification of total region of interest (ROI) in HER2high tumor and HER2low tumor. (C) The total ROI of collected tumors were quantified in HER2high and HER2low tumor tissue at 72 h (n = 2). Bars, SEM. | ||
αHER2/PLD improves the therapeutic efficacy in doxorubicin-sensitive and doxorubicin-resistant HER2-overexpressing tumors
Mice bearing 150–250 mm3 MCF7/HER2 or MDA-MB-361 xenografts were intravenously injected with 5 mg kg−1 αHER2/PLD, αDNS/PLD, PLD or saline. The mice were treated once a week for three weeks and the tumor size and body weight were measured once a week. αHER2/PLD significantly suppressed the growth of MCF7/HER2 tumors as compared to treatment with αDNS/PLD or PLD (P < 0.0001) (Fig. 7A). Importantly, in the doxorubicin-resistant tumor MDA-MB-361 model, αHER2/PLD delayed tumor growth more than PLD (P = 0.02) (Fig. 7B). There were no significant differences in the mean weight of mice treated with αHER2/PLD, αDNS/PLD or PLD (Fig. S4A and S4B†). Thus, αHER2/PLD enhanced the therapeutic efficacy of PLD against both doxorubicin-sensitive and doxorubicin-resistant tumors.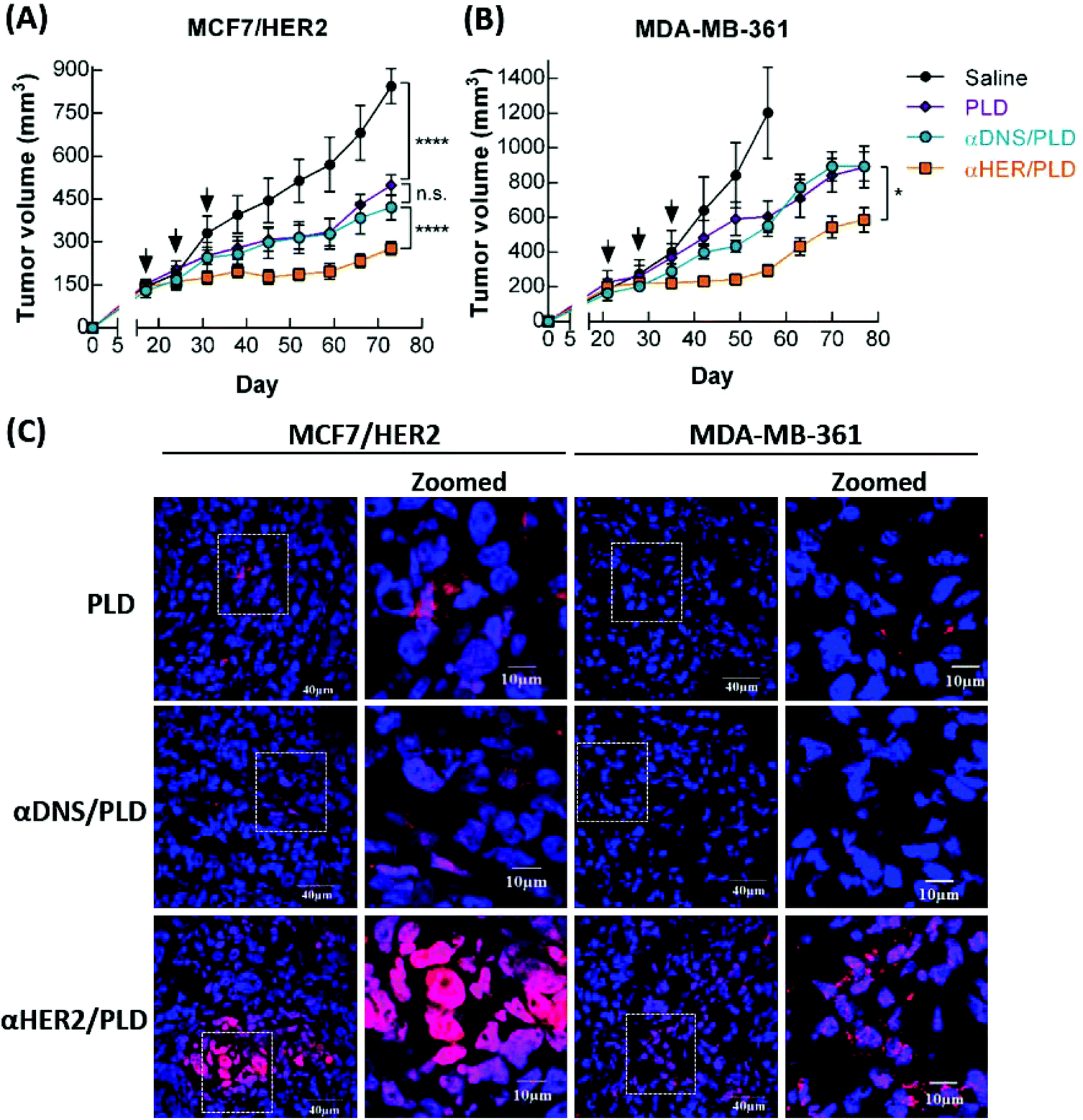 | ||
Fig. 7 The therapeutic efficacy and drug accumulation of αHER2/PLD in HER2-overexpressing tumors. Amounts of 5 mg kg−1 of αHER2/PLD ( ), PLD ( ), PLD ( ), αDNS/PLD ( ), αDNS/PLD ( ) or saline (●) were intravenously injected in mice bearing (A) MCF7/HER2 tumors and (B) MDA-MB-361 tumors in m.f.p. Treatment was performed once a week for 3 weeks. Results show the mean tumor size. Bars, SEM. Data were analyzed by repeated measures ANOVA. ****P < 0.0001, *P < 0.05; n.s., not significant. (C) The MCF7/HER2 (left) MDA-MB-361 (right) tumors were collected at 48 h and were stained with DAPI. The doxorubicin and DAPI fluorescence was detected by confocal microscopy. ) or saline (●) were intravenously injected in mice bearing (A) MCF7/HER2 tumors and (B) MDA-MB-361 tumors in m.f.p. Treatment was performed once a week for 3 weeks. Results show the mean tumor size. Bars, SEM. Data were analyzed by repeated measures ANOVA. ****P < 0.0001, *P < 0.05; n.s., not significant. (C) The MCF7/HER2 (left) MDA-MB-361 (right) tumors were collected at 48 h and were stained with DAPI. The doxorubicin and DAPI fluorescence was detected by confocal microscopy. | ||
To examine whether doxorubicin in αHER2/PLD accumulated in tumor cells, mice bearing either MCF7/HER2 or MDA-MB-361 tumors were intravenously injected with αHER2/PLD, αDNS/PLD or PLD, and then the tumors were collected at 48 h post-injection and tumor sections were stained with DAPI and the red fluorescence derived from doxorubicin was observed by confocal microscopy. Fig. 7C shows that more doxorubicin was detected in tumor cells in mice treated with αHER2/PLD as compared to mice treated with αDNS/PLD or PLD. A purple signal was noticeable, indicating the localization of doxorubicin to the nuclei of the tumor cells. In contrast, lower co-localization of doxorubicin and DAPI was observed in cancer cells in mice treated with PLD or αDNS/PLD. These results demonstrate that αHER2/PLD can enhance the accumulation of doxorubicin in the nuclei of HER2+ tumors that are sensitive or resistant to doxorubicin.
Toxicity of αHER2/PLD in human cardiomyocytes and mice
To examine possible doxorubicin-induced cardiotoxicity of αHER2/PLD, we used echocardiography to monitor cardiac function in BALB/c mice treated with αHER2/PLD. The mice were treated with 15 mg kg−1 αHER2/PLD, PLD, free doxorubicin or saline, and at 7 days post-treatment, the percentage of fractional shortening (FS) was measured, which is the degree of shortening of the left ventricular diameter between end-diastole and end-systole.35 A numerical value of FS that is lower than the normal baseline indicates reduced systolic function. Fig. 8A shows that the mean FS values for saline, free doxorubicin, PLD and αHER2/PLD are 42.3%, 27.6%, 36.4% and 37.8%, respectively; the mice treated with αHER2/PLD showed a similar numerical value to those treated with PLD. Next, we injected 5 mg kg−1 αHER2/PLD, PLD or saline in BALB/c mice once a week for 3 weeks to mimic a normal therapeutic schedule and then the mean FS was measured by echocardiography at 28 days post-first treatment. Fig. 8B shows that the mean FS values for saline, PLD and αHER2/PLD were 42.4%, 34.7% and 37.8%, respectively; the value for αHER2/PLD was slightly higher than that for PLD, but was not significantly different from that for PLD. The results indicate that αHER2/PLD might not lead to serious doxorubicin-induced cardiotoxicity in normal mice.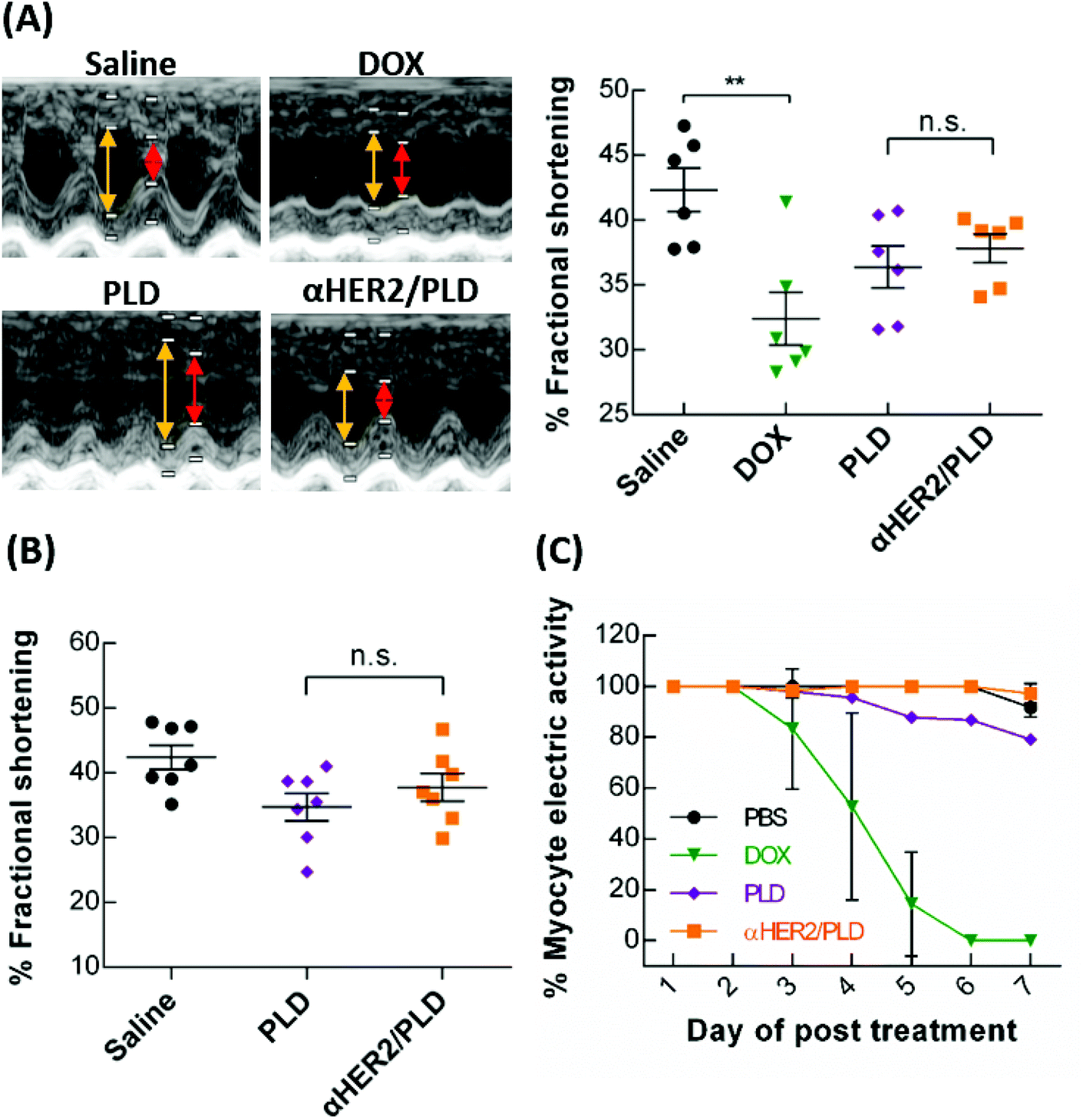 | ||
Fig. 8 αHER2/PLD did not enhance the cardiotoxicity of PLD. (A) BALB/c mice were treated with 15 mg kg−1 of doxorubicin ( ), PLD ( ), PLD ( ), αHER2/PLD ( ), αHER2/PLD ( ) or saline (●). The mean percentage of fractional shortening (FS) was determined by echocardiography of mice at 7 days post-treatment (n = 6). Yellow lines denote LVEDD and red lines denote LVESD. (B) BALB/c mice were treated with 5 mg kg−1 of PLD ( ) or saline (●). The mean percentage of fractional shortening (FS) was determined by echocardiography of mice at 7 days post-treatment (n = 6). Yellow lines denote LVEDD and red lines denote LVESD. (B) BALB/c mice were treated with 5 mg kg−1 of PLD ( ), αHER2/PLD ( ), αHER2/PLD ( ) or saline (●), once a week for 3 weeks and the percentage mean FS was determined by echocardiography (n = 7). (C) The hiPSC-CMs were treated with 1 μg mL−1 PBS (●), 1 μg mL−1 doxorubicin ( ) or saline (●), once a week for 3 weeks and the percentage mean FS was determined by echocardiography (n = 7). (C) The hiPSC-CMs were treated with 1 μg mL−1 PBS (●), 1 μg mL−1 doxorubicin ( ), 1 μg mL−1 PLD ( ), 1 μg mL−1 PLD ( ), or αHER2/PLD ( ), or αHER2/PLD ( ) for 7 days. The myocyte electric activity was recorded by MEA (untreated, n = 110; doxorubicin, n = 138; PLD, n = 117; αHER2/PLD, n = 156). Bars, SEM. Statistics indicate Student unpaired t test. **P < 0.01; n.s., not significant. ) for 7 days. The myocyte electric activity was recorded by MEA (untreated, n = 110; doxorubicin, n = 138; PLD, n = 117; αHER2/PLD, n = 156). Bars, SEM. Statistics indicate Student unpaired t test. **P < 0.01; n.s., not significant. | ||
Because human cardiomyocytes express low levels of HER2, the combination treatment with Herceptin and doxorubicin caused serious cardiotoxicity in clinical trials.36 Therefore, we examined whether mPEG × HER2 can induce cardiotoxicity in human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CM), which also express low levels of HER2.37 The hiPSC-CM cells were incubated with PBS, αHER2/PLD, PLD or free doxorubicin and then the beating cells were monitored on a microelectrode array. The percentage of myocyte electric activity was defined as the number of beating cells after treatment as compared to pre-treatment. Fig. 8C shows that doxorubicin induced a substantial decrease in myocyte electric activity after 4 days, while αHER2/PLD and PLD treatment retained at least 80% of myocyte electric activity after 7 days. The results indicate that the cardiotoxicity of αHER2/PLD was similar to that of PLD in hiPSC-CM.
To examine the general toxicity of αHER2/PLD, we analyzed the tissue sections of mice which were injected with repeat doses of αHER2/PLD, PLD and saline at 28 days post-first treatment. The results show αHER2/PLD did not cause serious side effects and response is similar to that of PLD (Fig. S5†). Collectively, these findings suggest that mPEG × HER2 did not synergistically induce the toxicity of PLD in mice.
Discussion
We successfully developed a BsAb (mPEG × HER2) that can simply and rapidly confer HER2 specificity to PEGylated drugs (e.g. PLD). Mixing mPEG × HER2 with PEGylated liposomal doxorubicin thus converted this clinically used medicine to a tumor-targeted drug (αHER2/PLD). Importantly, αHER2/PLD actively accumulated at and internalized into HER2-overexpressing cells, thus improving the therapeutic efficacy of PLD, which enhanced the susceptibility of breast cancer cells to doxorubicin. Moreover, the one-step and site-specific attachment of mPEG × HER2 to mPEG chains provides a simple approach to manufacture targeted PEG-NPs and maintain a stable and homogeneous antibody orientation. Thus, the BsAb can be modified on diverse mPEG-based materials, such as liposome, Qdot, FeOdot and AuNP33 and micelle.38 In addition, by designing a panel of BsAbs, a diverse set of PEG-NPs can be rapidly created to target different biomarkers (e.g., EGFR,39,40 ICAM-1,41etc.) expressed in individual diseases for diagnosis and therapy. We believe that mPEG BsAb overcomes many limitations of traditional manufacture of targeted PEG-NPs and expect that it will accelerate the development of targeted PEG-NPs for clinical use.It is important to induce the endocytosis of targeted NPs to enhance intracellular drug accumulation. Sapra and colleagues demonstrated that an internalizing PLD targeted to CD19 enhanced the life span of lymphoma-bearing mice by 2.86-fold relative to treatment with a non-internalizing PLD targeted to CD20.42 We previously reported that HCC36 cells that expressed internalizing anti-PEG antibody receptors were 3.48-fold more sensitive to PLD than HCC36 cells that expressed the same receptor that could not undergo endocytosis.43 In addition, accumulation of a PEGylated near-IR probe was 3.54-fold greater in HCC36 tumors that expressed endocytic receptors as compared to tumors expressing a non-endocytic receptor.43 In the current study, we found that αHER2/PLD increased the particle internalization and accumulation in lysosomes after 130 min incubation. This led to increased intracellular accumulation of doxorubicin in MCF7/HER2 cells. αHER2/PLD also displayed 3.0-fold increased specific targeting of Lipo-DiR in mice bearing MCF7/HER2 tumors as compared with MCF7/neo1 tumors; more importantly, αHER2/PLD exhibited enhanced doxorubicin accumulation in tumor nuclei and anti-tumor activity to MCF7/HER2 cells as compared with PLD (P < 0.0001). Thus, we demonstrated that mPEG × HER2 modification of PLD could endow PEG-NPs with the ability to undergo receptor-mediated endocytosis and active targeting in tumor cells to improve the therapeutic efficacy against HER2-overexpressing tumors. The results suggest that efficient internalization is a key direction for the development of targeted NPs.
Drug resistance is still one of the major limitations in the clinical treatment of breast cancer. The mechanisms behind resistance can be roughly divided into “acquired” and “intrinsic” resistance.44 Acquired mechanisms of resistance include the overexpression of the ATP binding cassette (ABC) transporter family (e.g. P-glycoprotein) that cause cells to fail to accumulate a drug through active drug efflux.45,46 In addition, a major intrinsic resistance mechanism is alterations in the TOP2A gene, a target of doxorubicin, which is deleted in more than 40% (23/57) of primary tumors with ErbB-2 amplification.11 Green et al. reported that patients treated with PLD who had tumors with low TOP2A expression levels had a shorter time to progression than those with high TOP2A levels.47 Likewise, reducing the tumor concentration of TOP2A protein by shRNA also decreased sensitivity to doxorubicin.48 Walker et al. proposed that changes of the cellular uptake or efflux of drugs are important factors to overcome resistance to doxorubicin.49 In our study, we focused on drug-resistant MDA-MB-361 breast cancer cells with a deletion of TOP2A, which have rarely been discussed in studies of targeted NPs. We found that αHER2/PLD significantly enhanced by 28-fold the cytotoxicity in MDA-MB-361 cells as compared to PLD. In addition, we observed that αHER2/PLD increased the nuclear accumulation of doxorubicin and significantly reduced tumor growth as compared with treatment of PLD (P < 0.05). Enhancing drug accumulation in tumor nuclei enhanced the susceptibility of TOP2A-deletion cancer cells to doxorubicin. Thus, targeted NPs can help overcome drug resistance via enhancing drug accumulation in tumor nuclei.
Irreversible acute and/or chronic cardiac dysfunction in a serious dose-limiting toxicity has been associated with doxorubicin.50 Co-treatment of patients with trastuzumab and doxorubicin increased the incidence of cardiac toxicity; thus this combination was not approved by FDA.51–53 Although PLD reduces doxorubicin-induced cardiotoxicity, Chia et al. reported that the co-treatment of PLD and trastuzumab produced some cardiotoxicity and/or asymptomatic declines in LVEF.54 We found in the present study that αHER2/PLD affected cardiac function to a similar extent to PLD and to a lower degree than doxorubicin in a mouse model with single or repeated dose treatment. αHER2/PLD also caused damage to human cardiomyocytes similar to that caused by PLD. These results are in agreement with Hendriks et al., who reported that HER2-targeted PLD does not enter human cardiomyocytes because the level of HER2 expressed by cardiomyocytes is insufficient to cause targeted NP binding and endocytosis.55 Thus, αHER2/PLD may induce less cardiotoxicity than the combination of doxorubicin and trastuzumab.
Experimental
Cell lines and animals
Expi293F cells (Thermo Fisher Scientific) were cultured in Expi293 expression medium (Thermo Fisher Scientific) on shakers (25 mm shaking diameter) with a shake speed of 120 rpm in a humidified atmosphere of 8% CO2 in air at 37 °C. Human breast cancer cells MCF7/HER2 (a clone stably transfected with human HER256) and MCF7/neo1 (a clone stably transfected with vehicle vector56) were generously provided by Dr M. C. Hung (Department of Molecular and Cellular Oncology, University of Texas, M. D. Anderson Cancer Center, Houston, TX). Doxorubicin-resistant breast cancer cells MDA-MB-361 (TOP2A gene deletion) were generously provided by TTY Biopharm Co. Ltd. MCF7/HER2-18, MCF7/neo1 and MDA-MB-361 cells were cultured in Dulbecco's Modified Eagle Medium/Nutrient Mixture F-12 (DMEM/F12, Gibco) medium supplemented with 10% (v/v) fetal bovine serum (FBS; Biological industries), 100 units per mL penicillin, and 100 μg mL−1 streptomycin (Gibco Laboratories) in a humidified atmosphere of 5% CO2 in air at 37 °C. hiPSC-CMs were derived from an unidentified healthy Taiwanese female patient as previously described.57 The hiPSC-CMs (at 30–40 days after differentiation) were further purified by culture in glucose-free-plus-lactate RPMI medium for 3–7 days. Healthy 6- to 8-week-old female BALB/c mice, BALB/c nude mice (BALB/cAnN.Cg-Foxn1nu/CrlNarl) and SCID mice (CB17/lcr-Prkdcscid/CrlNarl) were purchased from the National Laboratory Animal Center, Taipei, Taiwan. All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Kaohsiung Medical University and approved by the institutional animal care and use committee (IACUC) of Kaohsiung Medical University (IACUC number: 103005).Bispecific antibodies and nanoparticles
Human BsAbs were created by linking the C-terminus of an anti-mPEG Fab (clone h15-2b33) to an anti-HER2 scFv or anti-DNS scFv via a flexible peptide (GGGGS)3 to form mPEG × HER2 and mPEG × DNS, respectively. The anti-HER2 scFv was constructed by linking the 4D5 VH and VL domains with a linker (GGGGS)3. The anti-DNS scFv has been described previously.58 The VL-Cκ and VH-CH1-linker-scFv domains were separated with an IRES in the pLNCX retroviral vector (BD Biosciences, San Diego, CA) in the unique Hind III and Cla I restriction enzyme sites to generate pLNCXmPEG × HER2 and pLNCX-mPEG × DNS plasmids. Expi-293 cells were transfected with plasmids and the culture medium were collected after five days. The BsAbs were purified by affinity chromatography on gel prepared by reacting 36 mg of O-(2-aminoethyl)-O′-methylpolyethylene glycol 750 (Sigma Aldrich) per gram of CNBr-activated sepharose™ 4B (GE Healthcare, Little Chalfont, UK). Lipo/DOX® (PLD) was from TTY Biopharm Co. Ltd. Qdot655 nm (Qtracker® nontargeted quantum dots) was from Thermo Fisher Scientific. Lipo-DiR (PEGylated DOPC/CHOL liposomes labeled with DiR) was from FormuMax Scientific, Inc.Characterization of bispecific antibodies
mPEG × HER2 and mPEG × DNS (5 μg) were mixed with reducing or non-reducing loading dye, boiled for 10 min, and then electrophoresed on a 10% (w/v) SDS-PAGE gel and stained with Coomassie Brilliant Blue. Lane M is a PageRuler™ prestained protein ladder. Functional binding of the BsAbs was tested by sandwich ELISA. MCF7/HER2 cells (2 × 105 cells per well) were seeded overnight in 96-well plates (Nalge Nunc International, Roskilde, Denmark) coated with 50 μg mL−1 poly-D-lysine (Corning, New York) in culture medium at 37 °C. After extensive washing, the cells were fixed with 2% (w/v) paraformaldehyde for 5 min at room temperature (RT) and the reaction was stopped by the addition of 0.1 M glycine solution for 2 h at RT. Serial dilutions of mPEG × HER2 or mPEG × DNS were added to the wells at RT for 45 min. After extensive washing with PBS, 10 μg mL−1 of PLD was added to the wells for 20 min. After extensive washing with PBS, the bound PLD was determined by sequential addition of 10 μg mL−1 6-3 anti-PEG backbone antibody for 1 h, and 0.4 μg mL−1 goat anti-mouse IgG Fc-HRP (Jackson ImmunoResearch Laboratories). The wells were washed and 2,2′-azino-bis[3-ethylbenzothiazoline-6-sulphonic acid] (ABTS) and 30% hydrogen peroxide was added for 40 min before the absorbance values at 405 nm were measured with an EZ Read 400 ELISA (Biochrom). The kinetics of anti-PEG portion of mPEG × HER2 was analyzed by BLItz® system (ForteBio, Menlo Park, CA). The diluted concentration of mPEG × HER2 in PBS was added in the sensor, which immobilized to mPEG/BSA, and then the association and dissociation values of mPEG × HER2 were measured for each step of 78 s. The equilibrium dissociation constant was calculated from a nonlinear local fit of the data between mPEG × HER2 and mPEG/BSA, using the BLItz Pro 1.1 software.One-step formulation of mPEG-NPs with BsAbs
mPEG × HER2 and mPEG × DNS were mixed with PLD in PBS at 4 °C for 5 min to form αHER2/PLD and αDNS/PLD, respectively. αHER2/PLD was prepared with BsAb-to-mPEG molar ratios of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :360, 2:360, 4:360, 8:360 and 16:360. Based on a 100 nm liposome containing ∼80000 phospholipid molecules59 and ∼4528 mPEG-DSPE (the molar ratio of DSPC:cholesterol:DSPE-PEG2000 = 3:2:0.3), the corresponding number of BsAbs per PLD was estimated to be 13, 25, 50, 100 and 200, respectively. To quantify the free BsAb in αHER2/PLD, the αHER2/PLD with different ratios were incubated in mPEG/BSA-coated 96-well plate at RT for 45 min. After extensively washing the well, the BsAb was detected by goat anti-human Fab-HRP, and then ABTS substrate was added for 30 min before absorbance values at 405 nm were measured by an EZ Read 400 ELISA. mPEG × HER2 and mPEG × DNS were mixed with Qdot655 nm at 26 nmol BsAb per μmol Qdot to form αHER2/Qdot655 nm and αDNS/Qdot655 nm, respectively. mPEG × HER2 and mPEG × DNS were mixed with Lipo-DiR at a BsAb-to-phospholipid modified ratio of 1.4 nmol BsAb per mmol Lipo-DiR to form αHER2/Lipo-DiR and αDNS/Lipo-DiR, respectively.
:360, 2:360, 4:360, 8:360 and 16:360. Based on a 100 nm liposome containing ∼80000 phospholipid molecules59 and ∼4528 mPEG-DSPE (the molar ratio of DSPC:cholesterol:DSPE-PEG2000 = 3:2:0.3), the corresponding number of BsAbs per PLD was estimated to be 13, 25, 50, 100 and 200, respectively. To quantify the free BsAb in αHER2/PLD, the αHER2/PLD with different ratios were incubated in mPEG/BSA-coated 96-well plate at RT for 45 min. After extensively washing the well, the BsAb was detected by goat anti-human Fab-HRP, and then ABTS substrate was added for 30 min before absorbance values at 405 nm were measured by an EZ Read 400 ELISA. mPEG × HER2 and mPEG × DNS were mixed with Qdot655 nm at 26 nmol BsAb per μmol Qdot to form αHER2/Qdot655 nm and αDNS/Qdot655 nm, respectively. mPEG × HER2 and mPEG × DNS were mixed with Lipo-DiR at a BsAb-to-phospholipid modified ratio of 1.4 nmol BsAb per mmol Lipo-DiR to form αHER2/Lipo-DiR and αDNS/Lipo-DiR, respectively.
HER2 binding of BsAb/PLD
To examine the ability of PLD modified with various ratios of mPEG × HER2 to bind to cancer cells expressing HER2, MCF7/HER2 cells (2 × 105 cells per well) were seeded in poly-D-lysine-coated 96-well plate overnight at 37 °C. After fixing the cells, 625 ng mL−1 of αHER2/PLD made with densities of BsAb on PLD of 13, 25, 50, 100 and 200 BsAb/PLD were added to the wells at RT for 20 min. After extensive washing with PBS, the bound concentrations of PLD were determined by sequentially adding 10 μg mL−1 of 6-3 anti-PEG antibody for 1 h, washing with DMEM three times, and then adding 0.4 μg mL−1 of goat anti-mouse IgG Fc-HRP. The wells were washed three times with PBS and then ABTS substrate was added for 30 min before absorbance values at 405 nm were measured with an EZ Read 400 ELISA. To further analyze HER2 specific targeting efficacy of optimized BsAb-modified PLD, serial dilutions of αHER2/PLD and αDNS/PLD (with ratios of 100 BsAb/PLD) were incubated with MCF7/HER2 cells in poly-D-lysine-coated 96-well plates. PLD binding was measured as described above.Internalization of αHER2/PLD and intracellular accumulation of doxorubicin
Internalization of αHER2/PLD into HER2+ cancer cells was examined by adding 2 μg mL−1 of PLD, αDNS/PLD or αHER2/PLD in staining buffer (PBS containing 0.05% (w/v) bovine serum albumin) to 2 × 105 MCF7/HER2 cells for 40 min at 4 °C. After extensive washing with PBS, the cells were transferred to fresh culture medium and incubated for 0.5, 1 and 3 h at 37 °C. Control cells were incubated at 4 °C for 3 h. PLD on the surface of MCF7/HER2 cells was determined by sequential staining with 10 μg ml−1 6-3 anti-PEG antibody for 30 min and 4 μg ml−1 goat anti-mouse IgG Fcγ-FITC (Jackson ImmunoResearch Laboratories). After extensive washing with PBS, the fluorescence of cells was measured with a Cytomics FC500 flow cytometer. To examine the cellular accumulation of doxorubicin afforded by αHER2/PLD generated with various BsAb ratios, MCF7/HER2 cells (4 × 105 cells per well) were seeded in 6-well plates overnight at 37 °C. 1 μg mL−1 of PLD and αHER2/PLD with densities of BsAb on PLD of 13, 25, 50, 100 and 200 BsAb/PLD were added to the wells for 24 h at 37 °C. After extensive washing with PBS, the cells were suspended with 0.25% (v/v) trypsin/EDTA (Gibco). Then the doxorubicin fluorescence of cells was measured with a Cytomics FC500 flow cytometer (Beckman Coulter).Confocal live cell imaging
MCF7/HER2 cells (2 × 105 cells per well) were seeded on 2 μg mL−1 poly-L-lysine-coated glass slides in DMEM/F12 medium supplemented with 10% FBS (culture medium) at 37 °C in a humidified atmosphere of 5% CO2 in air for 24 h. The cells were incubated with 0.5 mg mL−1 Hoechst 33342 (Invitrogen) to stain nuclei and 50 nmol of LysoTracker Green DND-26 in RPMI (Sigma-Aldrich) to stain lysosomes for 40 minutes at 37 °C in an atmosphere of air containing 5% CO2. After washing with fresh culture medium, the cells were stained with 0.01 nmol of αHER2/Qdot655 nm and αDNS/Qdot655 nm in fresh culture medium at 37 °C and the fluorescence signals were recorded in real time with a Zeiss LSM780 laser-scanning microscope (Carl Zeiss AG).In vitro cytotoxicity
MCF7/HER2 cells (1 × 103 cells per well) were seeded in 96-well plates at 37 °C overnight. The cells were incubated with serial dilutions of PLD, αDNS/PLD or αHER2/PLD (100 μl per well) at 37 °C for 3 h. The medium was replaced with fresh medium and the cell viability was measured with the ATPlite™ luminescence assay system (PerkinElmer, Inc., Waltham, MA) after 96 h incubation. The same procedure was performed using 3 × 103 MCF7 per neo1 cells per well and 8 × 103 MDA-MB-361 cells per well for measuring in vitro cytotoxicity. Results are expressed as percent inhibition of luminescence as compared with untreated cells according to the following formula: %cell viability = 100 × (treated luminescence/untreated luminescence). The standard deviation for each data point was averaged over four samples.In vivo optical imaging
BALB/c nude mice bearing MCF7/HER2 (HER2high) and MCF7/neo1 (HER2low) tumors (∼100 mm3) in the mammary fat pad (m.f.p.) were intravenously injected with αHER2/Lipo-DiR, αDNS/Lipo-DiR, or Lipo-DiR (DiR concentration: 10 nmol per mouse). The mice were pentobarbital anesthetized and imaged with an IVIS spectrum optical imaging system (excitation, 750 nm; emission, 780 nm; PerkinElmer, Inc.) at 24, 48 and 72 h after injection. The MCF7/HER2 and MCF7/neo1 tumors were collected at 72 h. The ROIs in tumor areas were drawn and analyzed with Living Image software version 4.2 (Caliper Life Sciences). Nude mice were intravenously injected with Lipo-DiR, αDNS/Lipo-DiR and αHER2/Lipo-DiR, and then we sacrificed the mice and collected different organs (liver, spleen, heart, lung, ovary and kidney) for detecting the fluorescence intensity of Lipo-DiR with IVIS at 72 h post-treatment.In vivo nuclear accumulation of doxorubicin
BALB/c nude mice bearing MCF7/HER2 tumors and SCID mice bearing MDA-MB-361 tumors (∼200 mm3) in the m.f.p. were intravenously injected with 5 mg kg−1 of αHER2/PLD, αDNS/PLD or PLD. Tumors collected at 48 h were embedded in Tissue-Tek® OCT™ Compound (Sakura Finetek USA, Inc.) at −80 °C overnight. The nuclei were stained with DAPI fluoromount-G (Southern Biotech, Inc.) and the fluorescence of doxorubicin and DAPI was observed under an Olympus FluoView™ FV1000 confocal microscope (Olympus Imaging America Inc.)In vivo antitumor therapy
BALB/c nude mice were inoculated in their m.f.p. with 3 × 106 MCF7/HER2 cells in 100 μL of 1:1 PBS/matrigel (BD Biosciences) and the mice were subcutaneously injected with 2.5 μg β-estradiol 17-valerate in 50 μL sesame oil once a week. After tumors reached a volume of 150 to 250 mm3, mice were randomized into groups of 4 to 6 mice per group of equal average tumor volume and dosed intravenously with saline or 5 mg kg−1 of PLD, αHER2/PLD, αDNS/PLD once weekly for 3 weeks, for a total dose of 15 mg kg−1 doxorubicin. Tumor measurements were performed once a week using calipers, and tumor sizes were calculated using the equation: volume = (length × width2)/2. Mice were weighed once a week to examine treatment toxicity. The same therapeutic procedure as described above was used for SCID mice inoculated with 1 × 107 MDA-MB-361 cells.
Transthoracic echocardiography
To examine the single-dose doxorubicin-induced cardiotoxicity, BALB/c mice at the age of 7 weeks were evaluated by non-invasive echocardiography in basal condition. Mice (n = 6) were i.v. injected with saline, 15 mg kg−1 of PLD, αHER2/PLD, αDNS/PLD or doxorubicin as a positive control and echocardiography was conducted after 7 days. The left ventricular (LV) echocardiogram was assessed in both parasternal long-axis and short-axis views at a frame rate of 120 Hz. LV end-diastolic dimension (LVEDD) and LV end-systolic dimension (LVESD) were measured from the LV M-mode at the midpapillary muscle level. The following parameters were measured: LVEDD, LVESD, and percentage fractional shortening (%FS), calculated as [(LVEDD − LVESD)/LVEDD] × 100. To examine the repeat dose of doxorubicin-induced cardiotoxicity, BALB/c mice (n = 6) were injected with saline, 5 mg kg−1 of PLD or αHER2/PLD once a week for 3 weeks, and the echocardiography procedure as described above was used.Electrophysiological recordings
Purified hiPSC-CM cells were seeded on a matrigel-coated Muse 64-lead microelectrode array chip (M64-GLx MEA, Axion BioSystems, Inc.). The hiPSC-CMs cells were treated with PBS, 50 μg mL−1 of PLD or αHER2/PLD. The basal electrophysiological signals of the beating hiPSC-CM cells were recorded on the day before drug treatment. On the day of drug administration, the electrophysiological signals of hiPSC-CM cells were recorded at 10 min and 60 min after drug treatment. Then the electrophysiology was monitored once per day for 7 consecutive days. The beating cells were determined by the electrophysiological signals, which were analyzed by Axion BioSystems (Atlanta, GA, USA). The percentage of myocyte electric activity was defined as the number of beating cells after treatment as compared to pre-treatment.Conclusions
In summary, mPEG × HER2 provides a simple and stable one-step formulation for site-specific modification of PLD to increase drug internalization and the therapeutic efficacy of PLD in HER2-overexpressing tumors. Nevertheless, because of non-covalent modification, a proportion of mPEG × HER2 may dissociate from PLD during circulation in vivo. Enhancing the affinity of the anti-PEG fragment in mPEG × HER2 is desirable to strengthen the interaction of BsAb with PLD and may further increase the anticancer activity. The BsAbs described here possess potential advantages for targeted NP therapy including: (i) a novel and simple modification method to replace the traditional chemical conjugation. (ii) The changeable properties and universal applicability of BsAb can direct diverse PEG-NPs to different biomarkers expressed in individual diseases for diagnosis and therapy. (iii) BsAb modification can enhance the endocytosis of PEG-NPs to improve the anticancer effect, thus enhancing the susceptibility of breast cancer cells to doxorubicin. We believe that BsAbs (mPEG × markers) can expand the clinical application of PEG-NPs for treating drug-resistant tumors via improved tumor-specific targeting, internalization and therapeutic efficacy.Author contributions
Yi-An Cheng and I-Ju Chen contributed equally to this work. Yi-An Cheng designed and performed experiments reported in the paper, analyzed data and wrote the manuscript; I-Ju Chen helped with experiments, data analysis and contributed to manuscript editing; Yu-Cheng Su helped with confocal microscopy experiments, data analysis for endocytosis; Kai-Wen Cheng, Yun-Chi Lu and Wen-Wei Lin helped with data analysis and contributed to manuscript editing; Yuan-Chin Hsieh and Chien-Han Kao provided technical guidance with bispecific antibody design; Fang-Ming Chen provided the information of breast cancer clinical treatments and current challenges; Steve R. Roffler provided the concept and contributed to manuscript writing and editing; Tian-Lu Cheng provided the concept, experimental design and contributed to manuscript writing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the Ministry of Science and Technology, Taiwan (MOST 107-2320-B-037-024-MY3, MOST107-2320-B-037-028-MY2); the National Health Research Institutes, Taiwan (NHRI-EX108-10729EI); Academia Sinica, Taiwan (AS-TP-107-L11); the Program for Translational Innovation of Biopharmaceutical Development-Technology Supporting Platform Axis, Academia Sinica, Taiwan; and the KMU-KMUH Co-Project of Key Research (KMU-DK108002) and Research Foundation (KMU-Q108001) from Kaohsiung Medical University, Taiwan. This study is also supported partially by Kaohsiung Medical University Research Center Grant (Drug Development and Value Creation Research Center) (KMU-TC108A03).Notes and references
- Q. He, Z. Zhang, F. Gao, Y. Li and J. Shi, Small, 2011, 7, 271–280 CrossRef CAS PubMed.
- C. Cheng, P. Chu, K. Chuang, S. Roffler, C. Kao, W. Tseng, J. Shiea, W. Chang, Y. Su and B. Chen, Cancer Gene Ther., 2009, 16, 83 CrossRef CAS PubMed.
- D. Bobo, K. J. Robinson, J. Islam, K. J. Thurecht and S. R. Corrie, Pharm. Res., 2016, 33, 2373–2387 CrossRef CAS PubMed.
- M. C. Woodle and D. D. Lasic, Biochim. Biophys. Acta, 1992, 1113, 171–199 CrossRef CAS.
- A. C. Anselmo and S. Mitragotri, Bioeng. Transl. Med., 2016, 1, 10–29 CrossRef PubMed.
- J. S. Suk, Q. Xu, N. Kim, J. Hanes and L. M. Ensign, Adv. Drug Delivery Rev., 2016, 99, 28–51 CrossRef CAS PubMed.
- H. Maeda, H. Nakamura and J. Fang, Adv. Drug Delivery Rev., 2013, 65, 71–79 CrossRef CAS PubMed.
- M. E. O'Brien, N. Wigler, M. Inbar, R. Rosso, E. Grischke, A. Santoro, R. Catane, D. G. Kieback, P. Tomczak, S. P. Ackland, F. Orlandi, L. Mellars, L. Alland, C. Tendler and C. B. C. S. Group, Ann. Oncol., 2004, 15, 440–449 CrossRef PubMed.
- D. B. Kirpotin, D. C. Drummond, Y. Shao, M. R. Shalaby, K. Hong, U. B. Nielsen, J. D. Marks, C. C. Benz and J. W. Park, Cancer Res., 2006, 66, 6732–6740 CrossRef CAS PubMed.
- A. W. El-Kareh and T. W. Secomb, Neoplasia, 2005, 7, 705–713 CrossRef CAS PubMed.
- T. A. Järvinen, M. Tanner, V. Rantanen, M. Bärlund, Å. Borg, S. Grénman and J. Isola, Am. J. Pathol., 2000, 156, 839–847 CrossRef.
- J. C. Brase, M. Schmidt, T. Fischbach, H. Sültmann, H. Bojar, H. Koelbl, B. Hellwig, J. Rahnenführer, J. G. Hengstler and M. C. Gehrmann, Clin. Cancer Res., 2010, 1078–0432 Search PubMed . CCR-1009-2471.
- D. L. Iden and T. M. Allen, Biochim. Biophys. Acta, 2001, 1513, 207–216 CrossRef CAS.
- K. F. Pirollo and E. H. Chang, Trends Biotechnol., 2008, 26, 552–558 CrossRef CAS PubMed.
- C. Mamot, R. Ritschard, A. Wicki, W. Kung, J. Schuller, R. Herrmann and C. Rochlitz, J. Drug Targeting, 2012, 20, 422–432 CrossRef CAS PubMed.
- T.-L. Cheng, K.-W. Liao, S.-C. Tzou, C.-M. Cheng, B.-M. Chen and S. R. Roffler, Cancer Gene Ther., 2004, 11, 380 CrossRef CAS PubMed.
- X. Qian, X.-H. Peng, D. O. Ansari, Q. Yin-Goen, G. Z. Chen, D. M. Shin, L. Yang, A. N. Young, M. D. Wang and S. Nie, Nat. Biotechnol., 2007, 26, 83–90 CrossRef PubMed.
- R. Popovtzer, A. Agrawal, N. A. Kotov, A. Popovtzer, J. Balter, T. E. Carey and R. Kopelman, Nano Lett., 2008, 8, 4593–4596 CrossRef CAS.
- M. Satpathy, L. Wang, R. Zielinski, W. Qian, M. Lipowska, J. Capala, G. Y. Lee, H. Xu, Y. A. Wang and H. Mao, Small, 2014, 10, 544–555 CrossRef CAS PubMed.
- H. Lee, A. F. Shields, B. A. Siegel, K. D. Miller, I. Krop, C. X. Ma, P. M. LoRusso, P. N. Munster, K. Campbell and D. F. Gaddy, Clin. Cancer Res., 2017, 23, 4190–4202 CrossRef CAS PubMed.
- F. Meric-Bernstam, A. Johnson, E. E. I. Dumbrava, K. Raghav, K. Balaji, M. Bhatt, R. K. Murthy, J. Rodon and S. A. Piha-Paul, Clin. Cancer Res., 2018, 25, 2033–2041 CrossRef PubMed.
- Y. Tang, F. Soroush, Z. Tong, M. F. Kiani and B. Wang, Int. J. Nanomed., 2017, 12, 671–681 CrossRef CAS PubMed.
- A. S. Manjappa, K. R. Chaudhari, M. P. Venkataraju, P. Dantuluri, B. Nanda, C. Sidda, K. K. Sawant and R. S. Murthy, J. Controlled Release, 2011, 150, 2–22 CrossRef CAS PubMed.
- G. Bendas, A. Krause, U. Bakowsky, J. Vogel and U. Rothe, Int. J. Pharm., 1999, 181, 79–93 CrossRef CAS PubMed.
- V. P. Torchilin, J. Narula, E. Halpern and B. A. Khaw, Biochim. Biophys. Acta, 1996, 1279, 75–83 CrossRef.
- S. Salmaso and P. Caliceti, in Peptide and Protein Delivery, Elsevier, 2011, pp. 247–290 Search PubMed.
- D. F. Nellis, D. L. Ekstrom, D. B. Kirpotin, J. Zhu, R. Andersson, T. L. Broadt, T. F. Ouellette, S. C. Perkins, J. M. Roach, D. C. Drummond, K. Hong, J. D. Marks, J. W. Park and S. L. Giardina, Biotechnol. Prog., 2005, 21, 205–220 CrossRef CAS PubMed.
- L. Nobs, F. Buchegger, R. Gurny and E. Allemann, J. Pharm. Sci., 2004, 93, 1980–1992 CrossRef CAS PubMed.
- M. L. Geddie, N. Kohli, D. B. Kirpotin, M. Razlog, Y. Jiao, T. Kornaga, R. Rennard, L. Xu, B. Schoerberl, J. D. Marks, D. C. Drummond and A. A. Lugovskoy, mAbs, 2017, 9, 58–67 CrossRef CAS PubMed.
- X. Sun, G. Zhang, D. Patel, D. Stephens and A. M. Gobin, Ann. Biomed. Eng., 2012, 40, 2131–2139 CrossRef PubMed.
- P. Hajdu, A. A. Chimote, T. H. Thompson, Y. Koo, Y. Yun and L. Conforti, Biomaterials, 2013, 34, 10249–10257 CrossRef CAS PubMed.
- W.-W. Lin, Y.-C. Hsieh, Y.-A. Cheng, K.-H. Chuang, C.-C. Huang, C.-H. Chuang, I.-J. Chen, K.-W. Cheng, Y.-C. Lu and T.-C. Cheng, Anal. Chem., 2016, 88, 12371–12379 CrossRef CAS PubMed.
- C. H. Kao, J. Y. Wang, K. H. Chuang, C. H. Chuang, T. C. Cheng, Y. C. Hsieh, Y. L. Tseng, B. M. Chen, S. R. Roffler and T. L. Cheng, Biomaterials, 2014, 35, 9930–9940 CrossRef CAS PubMed.
- G. Fadaeian, S. A. Shojaosadati, H. Kouchakzadeh, F. Shokri and M. Soleimani, Iran. J. Pharm. Res., 2015, 14, 395 CAS.
- M. A. Mitry and J. G. Edwards, Int. J. Cardiol. Heart Vasc., 2016, 10, 17–24 Search PubMed.
- A. Seidman, C. Hudis, M. K. Pierri, S. Shak, V. Paton, M. Ashby, M. Murphy, S. J. Stewart and D. Keefe, J. Clin. Oncol., 2002, 20, 1215–1221 CrossRef CAS PubMed.
- S. Eldridge, L. Guo, J. Mussio, M. Furniss, J. Hamre 3rd and M. Davis, Toxicol. Sci., 2014, 141, 547–559 CrossRef CAS PubMed.
- C.-Y. Su, M. Chen, L.-C. Chen, Y.-S. Ho, H.-O. Ho, S.-Y. Lin, K.-H. Chuang and M.-T. Sheu, Drug Delivery, 2018, 25, 1066–1079 CrossRef CAS PubMed.
- C. B. Howard, N. Fletcher, Z. H. Houston, A. V. Fuchs, N. R. Boase, J. D. Simpson, L. J. Raftery, T. Ruder, M. L. Jones and C. J. de Bakker, Adv. Healthcare Mater., 2016, 5, 2055–2068 CrossRef CAS PubMed.
- J. Cui, Y. Ju, Z. H. Houston, J. J. Glass, N. L. Fletcher, S. Alcantara, Q. Dai, C. B. Howard, S. M. Mahler and A. K. Wheatley, Adv. Healthcare Mater., 2019, 1801607 CrossRef PubMed.
- J. T. Huckaby, C. L. Parker, T. M. Jacobs, A. Schaefer, D. Wadsworth, A. Nguyen, A. Wang, J. Newby and S. K. Lai, Angew. Chem., 2019, 58, 5604–5608 CrossRef CAS PubMed.
- P. Sapra and T. M. Allen, Cancer Res., 2002, 62, 7190–7194 CAS.
- K. H. Chuang, H. E. Wang, F. M. Chen, S. C. Tzou, C. M. Cheng, Y. C. Chang, W. L. Tseng, J. Shiea, S. R. Lin, J. Y. Wang, B. M. Chen, S. R. Roffler and T. L. Cheng, Mol. Cancer Ther., 2010, 9, 1903–1912 CrossRef CAS PubMed.
- T. H. Lippert, H.-J. Ruoff and M. Volm, Arzneimittelforschung, 2008, 58, 261–264 CAS.
- J. Cox and S. Weinman, Hepatic Oncol., 2016, 3, 57–59 CrossRef PubMed.
- J. A. Endicott and V. Ling, Annu. Rev. Biochem., 1989, 58, 137–171 CrossRef CAS PubMed.
- H. Green, O. Stål, K. Bachmeier, L. Bäcklund, L. Carlsson, J. Hansen, M. Lagerlund, B. Norberg, Å. Franzén and A. Åleskog, Cancer Lett., 2011, 313, 145–153 CrossRef CAS PubMed.
- D. J. Burgess, J. Doles, L. Zender, W. Xue, B. Ma, W. R. McCombie, G. J. Hannon, S. W. Lowe and M. T. Hemann, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9053–9058 CrossRef PubMed.
- J. Walker, C. Martin and R. Callaghan, Eur. J. Cancer, 2004, 40, 594–605 CrossRef CAS PubMed.
- S. E. Lipshultz, J. A. Alvarez and R. E. Scully, Heart, 2008, 94, 525–533 CrossRef CAS PubMed.
- K. R. Chien, N. Engl. J. Med., 2006, 354, 789–790 CrossRef CAS PubMed.
- L. Gianni, E. Salvatorelli and G. Minotti, Cardiovasc. Toxicol., 2007, 7, 67–71 CrossRef CAS PubMed.
- B. R. H. Bird and S. M. Swain, Clin. Cancer Res., 2008, 14, 14–24 CrossRef CAS PubMed.
- S. Chia, M. Clemons, L.-A. Martin, A. Rodgers, K. Gelmon, G. R. Pond and L. Panasci, J. Clin. Oncol., 2006, 24, 2773–2778 CrossRef CAS PubMed.
- B. S. Hendriks, S. G. Klinz, J. G. Reynolds, C. W. Espelin, D. F. Gaddy and T. J. Wickham, Mol. Cancer Ther., 2013, 12, 1816–1828 CrossRef CAS PubMed.
- G. Guo, T. Wang, Q. Gao, D. Tamae, P. Wong, T. Chen, W.-C. Chen, J. E. Shively, J. Y. Wong and J. J. Li, Oncogene, 2004, 23, 535 CrossRef CAS PubMed.
- J. Ma, L. Guo, S. J. Fiene, B. D. Anson, J. A. Thomson, T. J. Kamp, K. L. Kolaja, B. J. Swanson and C. T. January, Am. J. Physiol.: Heart Circ. Physiol., 2011, 301, H2006–H2017 CrossRef CAS PubMed.
- K. H. Chuang, C. M. Cheng, S. R. Roffler, Y. L. Lu, S. R. Lin, J. Y. Wang, W. S. Tzou, Y. C. Su, B. M. Chen and T. L. Cheng, Bioconjugate Chem., 2006, 17, 707–714 CrossRef CAS PubMed.
- D. Kirpotin, J. W. Park, K. Hong, S. Zalipsky, W. L. Li, P. Carter, C. C. Benz and D. Papahadjopoulos, Biochemistry, 1997, 36, 66–75 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary Fig. S1–S5. See DOI: 10.1039/c9bm00323a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |