DOI:
10.1039/C9BM00140A
(Paper)
Biomater. Sci., 2019,
7, 3204-3212
Immobilization approaches can affect protein dynamics: a surface-enhanced infrared spectroscopic study on lipid–protein interactions†
Received
27th January 2019
, Accepted 13th May 2019
First published on 31st May 2019
Abstract
The intrinsically disordered Parkinson disease protein α-synuclein (αS) performs conformational changes induced by intermolecular protein–protein as well as by protein-membrane interactions. Aggregation of αS is a hallmark for the disease, however the role of the membrane in the aggregation process still needs to be clarified. We used a surface-enhanced infrared absorption (SEIRA) spectroscopic approach to investigate the effect of lipid interactions on αS conformation. The near-field detection of SEIRA allows to study exclusively structural changes of immobilized αS with the advantage that the supernatant remains undetected and thus does not interfere with the spectral read-out. self-assembled monolayer (SAMs) of mixed NHS-PEG-SH linker and MT(PEG)4 spacer molecules were utilized to immobilize αS. The linker/spacer composition of the SAM was adjusted to prevent αS–αS interactions. Two different methods were applied for site-specific (C-terminal and N-terminal) αS immobilization. The immobilized protein was then exposed to lipid vesicles and SEIRA difference spectra were recorded to monitor the αS conformation over time. Irrespective of the used immobilization method, αS tethering hindered lipid-induced conformational changes. The spectra also indicate that a fraction of the immobilized αS eventually desorbs from the surface into the supernatant solution. Desorbed αS performs conformational changes and formation of β-structured aggregates is observed upon interaction with either lipid vesicles or supplementary αS. Our study demonstrates that αS aggregates only when the protein is free in solution and that surface immobilization procedures, commonly used in many analytical applications, can change the dynamic behavior of proteins thereby affecting protein structure and function.
Introduction
The intrinsically disordered α-synuclein (αS) is a physiologically abundant protein. However, aggregates of this 140-amino-acid protein are major components of inclusions (Lewy bodies) found in the brain of patients who show symptoms of Parkinson's disease (PD). The presence of these αS- and lipid-rich Lewy bodies suggests that aggregation of αS is driven by interactions with membranes. It has been shown that αS interacts with synaptic vesicles in vivo and its overexpression inhibits neurotransmitter release.1 Moreover, several in vitro studies revealed that αS interacts with lipids and membranes.2–6 It was found that the N-terminus of αS binds to negatively charged membranes thereby adopting α-helical structure,3,4,5,7–9,10,11 that αS aggregates in presence of membranes,4,9,12 and that membrane interactions accelerate the formation of β-structured aggregates compared to protein aggregation in solution.9,13 However, formation of aggregates and fibrils were also observed at high protein concentrations, even in the absence of membranes, due to enhanced αS–αS intermolecular interactions.14,15 Fourier transform infrared spectroscopy (FTIR) is a technique highly suitable to monitor conformational changes of amyloid proteins in aqueous solution and in interaction with lipids.14,16–19 Most often, attenuated total reflection (ATR)-FTIR is applied for in vitro analysis of protein conformation.20–23 With this technique, detection of the IR-active sample is feasible up to several hundred nanometers penetration depth (dp) of the IR evanescence field into the sample placed on the internal reflection element (IRE) of the ATR unit.16 Recently, we demonstrated successful application of ATR-FTIR spectroscopy to study the interactions of αS with solid supported lipid bilayers (SSLB) as biomimetic membranes.9 A SSLB was formed on the IRE (also referred to as ATR crystal), and subsequent exposure of the SSLB to αS in solution facilitated the investigation of αS–membrane interactions. Since we used a silicon (Si) ATR crystal (dp ≈ 850 nm at 1000 cm−1), the IR evanescent field detected the protein–membrane interactions at the interface, but in addition the protein–protein interactions in the supernatant (Fig. 1). Hence conclusive detection of conformational changes, induced solely by protein–lipid interactions, becomes illusive. Moreover, sedimentation of αS on the lipid bilayer increases the protein concentration close to the SSLB, which in turn leads to more protein–protein interactions, and thus the αS–αS interactions in the supernatant will contribute to the aggregation of αS. One can speculate that under physiological conditions (low αS concentration) conformational changes of αS are mainly driven by the membrane, but this hypothesis is difficult to examine with conventional ATR because the protein in the supernatant and the protein in direct lipid contact will both contribute to the IR absorbance signal. To investigate exclusively the contribution of lipid interaction to the aggregation process of αS, intermolecular αS–αS interactions have to be prevented. Therefore, we explored here a novel methodological approach, namely SEIRA detection combined with site-specific (N- or C-terminal) immobilization of αS on a gold surface and subsequent exposure to lipid vesicles. The near-field effect of SEIRA offers the advantage to monitor solely the interactions between the immobilized αS monomers and the membrane without interference from αS in the supernatant solution (bulk αS). As compared to conventional ATR, the penetration depth of the IR beam into the sample is considerably less for SEIRA (dp < 10 nm) and thus does not or only negligibly detect the supernatant solution. In addition, the signal is enhanced in the vicinity of the metal surface which allows to detect samples with low concentrations.24,25 Lipid-induced conformational changes of immobilized αS were probed by adding POPG lipid vesicles to the supernatant. It has been shown that the affinity of αS to POPG vesicles is even higher compared to its affinity to lipid mixtures present under pathophysiological conditions26 and thus POPG vesicles are well suited for our proof-of-principle study. The high sensitivity of the SEIRA technique assures the detection of slightest changes in the structure of the immobilized αS monolayer induced by lipid interactions, while the lipid vesicles and the supernatant are not detected.
 |
| | Fig. 1 Schematic comparison of ATR and SEIRA experiments for investigation of αS–membrane interactions. (a) ATR experiment with a lipid bilayer formed on the surface of a Si IRE, and addition of αS in aqueous solution; a higher penetration depth (dp) of the evanescent IR field leads to simultaneous detection of protein–membrane interactions at the interface and αS–αS interactions in the supernatant solution. (b) SEIRA experiment with αS immobilized on a SAM of mixed PEG linkers (black) and spacers (violet). The lower penetration depth (dp) of the IR signal in the vicinity of the gold film results in the exclusive detection of the immobilized protein while the bulk solution is practically outside of the evanescent field. High surface sensitivity of SEIRA experiments allows for observation of slightest αS conformational changes. Size scales and proportions are not accurate and do not represent the reality. | |
Experimental section
SEIRA experiments
SEIRA measurements were performed by utilizing a single reflection unit using a Si prism IRE as reported before.27 A thin SEIRA-active film with gold nanostructures was deposited on a Si crystal by application of a deposition method described in details elsewhere.27–29 In brief, the surface of the Si prism was polished and covered with 40% w/v NH4F for 1 min to remove the residual oxide layer and to terminate the surface with hydrogen. Afterwards the Si prism was rinsed with water and tempered at 60 °C for 20 minutes. A 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 mixture of NaAuCl4 (0.03 M), and Na2SO3 (0.3 M) + Na2S2O3 (0.1 M) + NH4Cl (0.1 M), and HF (2% w/v) was deposited on the surface of the Si crystal for 1 minute. The crystal was rinsed with water. Possible contamination from plating was electrochemically removed by applying a dc voltage of +1.5 V between the gold thin film and a Pt counter electrode for 1 min in H2SO4 (0.1 M). SEIRA measurements were performed with a Vertex 80v FTIR spectrometer (Bruker). IR spectra were recorded with 100 scans, a resolution of 4 cm−1 and atmospheric compensation was applied. Fourier transformation was performed with a Mertz phase correction and a Blackman-Harris 3-term apodization. All SEIRA experiments were performed as difference measurements and thus are indicated as surface-enhanced infrared difference absorption (SEIDA) spectra.
:1:1 mixture of NaAuCl4 (0.03 M), and Na2SO3 (0.3 M) + Na2S2O3 (0.1 M) + NH4Cl (0.1 M), and HF (2% w/v) was deposited on the surface of the Si crystal for 1 minute. The crystal was rinsed with water. Possible contamination from plating was electrochemically removed by applying a dc voltage of +1.5 V between the gold thin film and a Pt counter electrode for 1 min in H2SO4 (0.1 M). SEIRA measurements were performed with a Vertex 80v FTIR spectrometer (Bruker). IR spectra were recorded with 100 scans, a resolution of 4 cm−1 and atmospheric compensation was applied. Fourier transformation was performed with a Mertz phase correction and a Blackman-Harris 3-term apodization. All SEIRA experiments were performed as difference measurements and thus are indicated as surface-enhanced infrared difference absorption (SEIDA) spectra.
SEIDA spectra of αS in solution on a non-modified gold film
SEIDA spectra of αS in solution (1 mg mL−1) were monitored on a bare gold film in order to investigate the potential influence of the gold film on the vibrational modes. The spectra were used as reference for band assignment of the SEIDA spectra with immobilized αS.
Self-assembled monolayers (SAMs) for αS immobilization
One possibility to assure that αS is in the vicinity of the SEIRA metal film is to tether the biomolecule to the surface. Immobilization can be achieved by modification of the gold layer with self-assembled monolayers (SAMs) of linker molecules.27,30–32 SAMs are considered to provide a stable and reproducible approach for the immobilization of molecules. SAMs of polyethylene glycol (PEG) have been utilized in several studies to modify a surface for specific protein immobilization.31,33 Thiol tail groups are suitable to link SAMs on gold films whereas functional head groups of SAMs can be applied for the selective immobilization of biomolecules.34–37 In a previous SEIRA study, we have devised SAMs of mixed NHS-PEG-SH (10 kDa) (NHS: N-hydroxysuccinimide) and MT(PEG)4 (methyl- and sulfhydryl-terminated PEG) for immobilization of poly-L-lysine (PLL) to gold.27 In this work, we used SAMs of 5 kDa NHS-PEG-SH (Nanocs) and MT(PEG)4 (Thermo Scientific), thereby we reduced the length of the NHS-PEG-SH linker molecules to ≈30 nm and facilitated protein immobilization closer to the gold surface leading to more enhancement of the absorbance signal. MT(PEG)4 are only 1.58 nm long, and were applied as inactive spacers to passivate the gold surface against non-specific interactions between αS and the gold film. In order to avoid uncontrolled αS aggregation caused by intermolecular interactions among the immobilized αS monomers, we reduced the number of NHS sites, available for protein immobilization, by adjusting the relative concentration of NHS-PEG-SH (linker) and MT(PEG)4 (spacer). Increasing the concentration of the MT(PEG)4 spacers in the mixed PEG SAM leads to an increased distance between the immobilized αS monomers which in turn decreases intermolecular interactions. Formation of mixed SAMs succeeded by exposure of the gold film to a 1 mM aqueous solution of mixtures of NHS-PEG-SH and MT(PEG)4 for ≈24 h with different linker:spacer compositions, i.e. 1:1, 1:10, and 1:100. Thus the SAM composition was adjusted by utilizing short spacers to passivate the gold surface and furthermore to create sufficient spacing among the longer linker molecules. The spacing prevented intermolecular interactions of the immobilized αS and subsequent exposure to lipid vesicles facilitated the investigation of αS conformational changes specifically induced by the lipids.
Immobilization of αS
The SAM of mixed PEGs was modified for protein immobilization. We used human α-synuclein recombinantly expressed in Escherichia coli and N-terminally tagged with histidine (Sigma-Aldrich). The protein was tethered at one of its two termini, either by modification of the SAM with an αS antibody (anti-synuclein-α antibody from rabbit, Sigma Aldrich) for C-terminal immobilization or with aminonitrilotriacetic acid (ANTA) for N-terminal tethering. Both procedures do not use covalent binding of αS to the surface and should allow for less surface confinement.38 For C-terminal immobilization, the SAM was exposed to an antibody solution (0.1 mg mL−1 in PBS buffer with 0.1 M sodium phosphate, 0.15 M NaCl, pH 7.2). The NHS ester head group of NHS-PEG-SH linker reacts with the amino group of the antibody. A similar procedure has been reported before for the immobilization of the Abeta peptide with an antibody, using a Germanium surface modified with triethoxysilanes and N-hydroxysuccinimidyl ester (NHS) linkers.22 After the reaction, the supernatant was removed and the surface was rinsed substantially with H2O to remove remaining free antibody. The PEG SAM was then modified with covalently bound antibodies. A background spectrum was recorded. Afterwards, αS (1 mg mL−1) was added to the supernatant and SEIRA difference spectra (SEIDA spectra) were taken for 2 hours following the (non-covalent) immobilization process of αS to the antibody. The surface was again rinsed with H2O to remove non-specifically adsorbed αS and αS in solution. SEIDA spectra were recorded over time to ensure the stability of the immobilized αS. Fig. 2a sketches the different steps of αS immobilization via the C-terminus.
 |
| | Fig. 2 Immobilization of αS to a NHS-PEG-SH:MT(PEG)4 SAM on a gold surface. Two different methods were applied for affinity-based, non-covalent immobilization of αS (a) C-terminal tethering of αS to an antibody modified SAM. (b) N-Terminal tethering of αS via His-tags to an ANTA modified SAM. | |
N-Terminal immobilization of αS was performed with an alternative and widely applied approach, namely affinity binding of the αS histidine-tags to surfaces modified with aminonitrilotriacetic acid (ANTA).39,40 A SAM of mixed PEGs was formed on the thin gold film. Afterwards the NHS-PEG-SH linker molecules were modified by exposure of the PEG SAM to ANTA as described in details elsewhere.39 In brief, the PEG SAM was rinsed with water and consecutively with potassium carbonate buffer (500 mM, pH 9.8). Then 2 mM ANTA solution (500 mM potassium carbonate buffer, pH 9.8) was added. The SAM was modified with ANTA overnight. Afterwards, the surface was rinsed with H2O and the supernatant was exchanged with protein-binding buffer (50 mM Tris, pH 7.4, 100 mM NaCl, 1 mM NiCl2, 1 mM MgCl2). A background spectrum was recorded, αS (1 mg mL−1) was added and the αS immobilization process was monitored with SEIDA spectra. Fig. 2b schematically depicts the steps of αS immobilization via the N-terminus.
Addition of lipid vesicles
To investigate the interactions of immobilized αS with the membrane, the protein was exposed to small unilamellar vesicles (SUVs) of POPG (1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-rac-(1′-glycerol)) and SEIDA were recorded over time. POPG lipids (Avanti Polar Lipids) were used without further processing. The desired amount was taken from the lipid stock solution to obtain a vesicle concentration of 2.5 mg mL−1. Chloroform was removed by placing the sample under a gentle stream of nitrogen for about 10 minutes. The resulting lipid film was placed in a vacuum chamber for two hours, resuspended in 1 mL Tris-HCl buffer (10 mM, pH 7.4), mixed thoroughly, and incubated at room temperature for 20 minutes. The SUVs were prepared by extrusion with a handheld extruder (Avanti Polar Lipids) through a filter-supported 30 nm polycarbonate membrane. When added to the supernatant, the lipid vesicles were close enough to interact with the immobilized αS as we verified by the rise of lipid signals at ∼2850 cm−1 and ∼2930 cm−1 in the spectra.
Control experiments with SEIRA and ATR
We performed SEIRA control experiments with supplementary αS (1 mg mL−1 in Tris-HCl buffer) which was added to the supernatant. Supplementary αS results in an increase of the local protein concentration in proximity of the immobilized αS and thus enhanced protein–protein interactions. Spectra were recorded over time. As mentioned above, the near-field nature of SEIRA restricts the detection of conformational changes to the immobilized αS monolayer, while the supernatant with αS in solution remains practically undetected. Thus the effect of additional protein interactions on the immobilized monomers could be tracked. Furthermore, ATR-FTIR control measurements of the supernatant solution were performed with a multi-reflection silicon ATR-cell (Bio-ATR II with 7–9 reflections, Bruker) and a Vertex 70v FTIR spectrometer (Bruker) equipped with a mercury cadmium telluride (MCT) detector. IR spectra were recorded with 32 scans and a resolution of 4 cm−1. Atmospheric compensation was applied to the spectra. The supernatant was removed after each SEIRA experiment and placed on the ATR crystal. Depending on the experiment, the supernatant comprised desorbed αS together with POPG SUVs or desorbed αS together with supplementary αS. The ATR control measurements provided insights into conformational changes and aggregation behavior of αS in the supernatant solution not detectable by SEIRA.
Results and discussion
Effect of the gold surface
Protein immobilization on gold surfaces might influence IR band frequencies when SEIRA spectra are compared to conventional ATR measurements.27,41 Thus we used SEIRA spectra of αS in solution placed directly on a non-modified gold film (Fig. 3) as reference for our studies with immobilized αS. A spectrum of H2O was recorded before addition of the protein solution. Difference spectra were taken and show the presence of the protein by the rise of the amide I (1648 cm−1–1659 cm−1) and amide II (≈1550 cm−1) bands. The amide I band at 1648 cm−1 (Fig. 3, 0 min) indicates a disordered conformation of αS in solution at the beginning of the experiment. Protein sedimentation on the gold surface and possible formation of αS multilayers leads to an intensity increase and a shift of the amide I band to 1656 cm−1 within a few minutes (Fig. 1, 5 min) and to 1659 cm−1 after hours. The band shift indicates that αS in solution performs a conformational change and adopts a more α-helical structure on the gold surface over time. The significant influence of surface interaction on protein conformation becomes obvious. It is most likely so prominent for αS, because the intrinsically disorderd structure maximizes the available contact surface.
 |
| | Fig. 3 SEIDA spectra of αS in aqueous solution on a non-modified bare gold film. Upon exposure to a gold layer, the spectrum shows a broad amide I band with a peak at 1648 cm−1 tentatively assigned to the disordered structure of αS. The spectrum was recorded a few seconds after αS addition and is depicted as 0 min. The amide I peak shifts to 1656 cm−1 and finally to 1659 cm−1. This shift indicates αS conformational changes from a disordered structure to a predominantly α-helical structure over time. | |
Optimization of SAM composition with linker and spacer molecules
SAMs of mixed PEGs were utilized for a controlled αS immobilization on the gold film.27,38,42 Our objective was to hinder intermolecular interactions among the immobilized αS monomers. Intuitively, lower relative concentration of linker molecule NHS-PEG-SH compared to the unreactive MT(PEG)4 spacer leads to less available binding sites for αS. We performed experiments with mixed SAMs and varied the linker:spacer compositions, 1:1, 1:10, and 1:100. Our experiments show that a lower linker concentration leads to immobilization of fewer αS molecules on the SAM as depicted exemplarily by Fig. S1, ESI.† This is manifested by the reduced intensity of the amide I and amide II bands of the αS immobilized to the NHS-PEG-SH:MT(PEG)4 1:100 SAM compared to the 1:1 SAM. SEIDA spectra demonstrate that a decrease of available binding sites reduces the protein binding. However, the SEIDA signal for αS immobilized on 1:1 SAM (Fig. S1a†) is only ∼3 times higher than for 1:100 SAM (Fig. S1b†). A possible explanation could be that the comparatively large sequence length of immobilized αS monomers cover the SAM substantially, and block a fraction of available linker molecules as depicted schematically in Fig. S2.† Thus, not all available linker molecules can bind αS monomers. As result from our experiments, we observe that the correlation between the number of available linker molecules and the effectively used immobilization sites is not linear. Also other effects might contribute to this nonlinearity, as the composition of the deposited SAM strongly depends on the surface affinity of the involved molecules and on the deposition time. Since αS immobilization to a 1:100 SAM is stable and reproducible, and intermolecular αS–αS interactions are reduced due to sufficient spacing between the immobilized αS monomers, we have chosen the 1:100 mixed SAM as the default SAM composition for all measurements in this work.
C-Terminal immobilized αS
For C-terminal immobilization, the 1:100 SAM was first functionalized with an αS C-terminal antibody as explained in the Experimental section. A SEIDA spectrum of the PEG SAM functionalized with the covalently bound antibody is shown in Fig. S3a (ESI†). Successful C-terminal immobilization of αS was monitored in Fig. S4 (ESI†) and the amide I band (≈1652 cm−1) hints a predominant disordered protein for the immobilized αS. The absorbance of the antibody does not contribute significantly to the total amide I signal of the immobilized protein (Fig. S5†). Membrane interactions were investigated by exposure of the immobilized protein to POPG vesicles, and SEIDA spectra were recorded over 24 hours. As depicted in Fig. 4a, the interaction of POPG SUVs with immobilized αS doesn't result in any significant spectral changes. Thus POPG vesicles did not induce any conformational changes to the αS monolayer immobilized at the C-terminus, in contrast to αS in solution where N-terminal conformational changes to α-helical structure have been observed after interaction with POPG SUVs.4,9,26 This observation is surprising since the immobilization at the C-terminus seems not to restrict the conformation and αS remains disordered after C-terminal immobilization similar to αS in solution. Thus the N-terminus should be accessible for vesicle interaction. The N-terminal amino acids 6–97 were reported to be involved in lipid interactions.26 Since the C-terminal antibody utilizes amino acids 91–140 (immunogen range) for binding the protein, most of the N-terminal amino acid residues (1–90) remain accessible after immobilization. One could hypothesize that direct interactions with the gold surface might have hindered conformational changes. However, we exclude αS interactions with gold since the MT(PEG)4 spacer molecules passivate the gold film and prevent protein interactions. The non-changing amide I band of the immobilized protein at ≈1650 cm−1 indicates that αS remains disordered even after long exposure to POPG SUVs (Fig. 4a). Thus, even carefully designed C-terminal immobilization of αS seems to constrain the conformational dynamics required for membrane interaction.
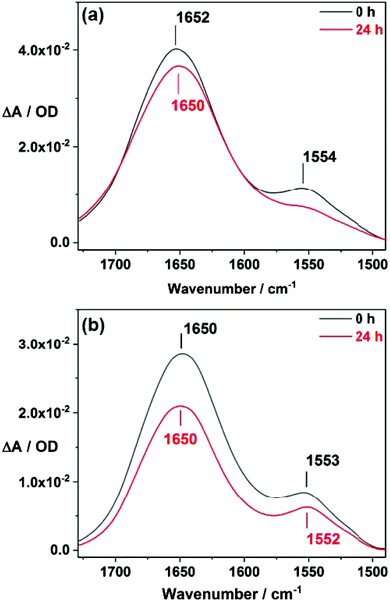 |
| | Fig. 4 Conformation of C-terminal immobilized αS monitored by SEIDA. A decrease in amide band intensities indicates desorption of immobilized αS from the surface over time. (a) Interaction with POPG SUVs. The disordered structure remains conserved for 24 h, lipid-induced conformational changes were not observed. (b) Effect of supplementary αS. No conformational changes were detected upon increase of protein–protein interactions. | |
Since the space between the monomers was adjusted to avoid protein interactions, immobilized αS monomers are not expected to aggregate. In order to validate the effect of αS–αS intermolecular interactions, supplementary αS was added to the supernatant solution. An increase of intermolecular interactions in the vicinity of the immobilized protein was expected since αS in solution sediments and interacts with the immobilized protein. SEIDA spectra were recorded to monitor this process (Fig. 4b). Again we have to emphasize that the immobilized αS predominantly contributes to the SEIDA signal whereas αS in the supernatant is detected only marginally. The spectra reveal that even increased intermolecular interactions do not trigger any significant conformational changes of the C-terminal immobilized αS. The amide I band at ≈1650 cm−1 hints that the immobilized αS remains disordered after several hours of interaction with supplementary αS. The spectra of C-terminal immobilized αS, before addition of either POPG vesicles or supplementary αS in the supernatant solution, are shown Fig. S6† and do not change compared to the spectra measured directly after addition (Fig. 4, 0 h).
N-Terminal immobilized αS
Immobilization of αS via N-terminus was achieved by utilizing the reaction of N-terminal histidine tags of the αS with the ANTA modified NHS-PEG-SH:MT(PEG)4 1:100 SAM. Fig. S7 (ESI†) shows the SEIDA spectra of N-terminal immobilized αS. After protein immobilization, the surface was rinsed with H2O to remove unspecifically adsorbed protein. The peak of the absorbance spectra at 1661 cm−1 shows a considerable shift to higher wavenumbers, compared to the C-terminal immobilized αS. This indicates that αS adopts a more α-helical structure after N-terminal immobilization, similar to the observed α-helical structure when αS is adsorbed to a non-modified gold film. We conclude that not only the interaction with membranes, but also the interaction of the αS N-terminus with surfaces can induce conformational changes.
N-Terminal immobilized αS was exposed to POPG SUVs to study lipid–protein interaction. SEIDA spectra of the N-terminal immobilized αS were recorded for 24 hours (Fig. 5a). A slight shift of the amide I absorbance peak to 1663 cm−1 was observed. However, we cannot conclude that the immobilized protein performs significant conformational changes, αS rather preserves the α-helical structure after long interaction with POPG SUVs. Thus N-terminal immobilization seems also to hinder further conformational changes of αS. Similar to the experiments for the C-terminal immobilized αS, supplementary αS was added to the supernatant solution. Again, the aim was to increase the αS concentration in proximity to the immobilized protein and thus to increase the intermolecular αS–αS interactions. SEIDA spectra do not indicate any conformational changes for the N-terminal immobilized αS (Fig. 5b). Hence, the reduced conformational degree of freedom is independent of the immobilization site and of the alternatively applied immobilization approach. Fig. S8† depicts the spectra of N-terminal immobilized αS before addition of either POPG vesicles or supplementary αS, and demonstrate again that the spectra do not change if measured directly after addition (Fig. 5, 0 h).
 |
| | Fig. 5 (a) Conformation of N-terminal immobilized αS monitored by SEIDA. Partial desorption of immobilized αS is reflected by the decrease of amide band intensities over time. (a) Interaction with POPG SUVs. After immobilization, αS adopts already a predominantly α-helical structure, however lipid interactions do not trigger further conformational changes. (b) Effect of increased protein–protein interactions. A higher αS concentration in the environment of the N-terminal immobilized protein does not lead to conformational changes. | |
Interactions of desorbed αS
Utilizing the protein's affinity to an antibody or His-tag for immobilization is not as stable as immobilization performed by covalent binding. Thus, a fraction of the immobilized αS will finally desorb from the surface over the course of the experiment (24 h), as shown in Fig. 4 and 5 by the decrease in band intensities of the amide bands. As a result, the supernatant will comprise desorbed αS besides POPG SUVs. Since our SEIDA experiment does not probe the supernatant, we analyzed it in a separate ATR-experiment. Therefore, at the end of the SEIDA experiments (after 24 h), 100 μL of the supernatant were removed and subsequently used in an ATR (control) experiment. ATR-FTIR spectra of the supernatant solution are shown in Fig. 6 exemplarily for αS desorbed from the C-terminal immobilized protein. It should be noted that IR band positions might be shifted when SEIRA- and ATR-spectra are compared because of the influence of the metal surface in an SEIRA experiment. However, it becomes obvious that the αS conformation in the supernatant solution is different from that in the immobilized protein. Several amide I band components occur in the ATR-spectra, in contrast to the SEIDA-spectra in Fig. 4 and 5. The various amide I components reflect the structural heterogeneity of the desorbed αS in solution after long (>24 h) interaction with POPG vesicles. Amide I band components at 1618 cm−1, 1634 cm−1, 1672 cm−1, and 1684 cm−1 hint that the desorbed αS forms β-sheets and β-structured aggregates upon interaction with POPG SUVs. This is in agreement with several studies that demonstrate αS aggregation upon interaction with lipids.27,43–46 Thus C-terminal immobilization of αS seems to inhibit lipid-induced protein conformational changes (Fig. 4a), while the desorbed “free” αS adopts a conformational heterogeneous structure, even to β-structured aggregates (Fig. 5a).
 |
| | Fig. 6 ATR-FTIR measurements of desorbed αS. (a) Interaction with POPG SUVs. (b) Effect of increased protein concentration. Both lipid–protein and protein–protein interactions induce conformational changes with a major fraction of β-sheets and β-structured aggregates. The supernatant solutions were taken from the SEIRA experiments after ≈24 h. | |
We also probed the effect of increased protein–protein interactions on desorbed αS. Therefore, supplementary αS (instead of POPG vesicles) was added to the supernatant of the SEIDA experiment. Afterwards (24 h), 100 μL of the supernatant was removed and subsequently analyzed by ATR measurements. The ATR-FTIR spectrum of the supernatant (Fig. 6b) reveals several amide I components at 1611 cm−1, 1632 cm−1, and 1684 cm−1, indicating that αS in solution also forms β-structured aggregates. Thus our ATR experiments of the supernatant proved that αS in solution performs conformational changes, with and without membrane interactions, in agreement with several other studies.8,9,14,44,45,47–50 In contrast, the immobilization of αS seems to prevent conformational dynamics. Reduced biological activity of covalently bound αS due to surface confinement was reported before.38 However, our SEIDA experiments clearly indicate that even non-covalent immobilization has a profound effect on the αS properties. This is manifested by the hindered ability of the immobilized αS to adopt α-helical structure in interaction with POPG SUVs, as well as the hindered conformational changes to form β-structured aggregates after exposure to supplementary αS. Only after desorption from the surface, αS formed β-structured aggregates, even without membrane interactions and similar to “free” αS in solution.
Conclusion
Application of the SEIRA technique allowed us to differentiate the effects of membrane versus protein interactions on αS aggregation. SEIRA is ideally suited because conformational changes of immobilized αS after interaction with POPG vesicles can be monitored without interference of the supernatant. We have utilized two different and well-established methods for site-specific immobilization of αS at both termini. Independent of which terminus of αS was immobilized or which immobilization procedure was used, no conformational changes were induced, neither after membrane interaction nor after increased protein–protein interactions. The conformational degree of freedom seems to be constrained for immobilized αS as compared to αS in solution. However, complementary studies show that αS aggregates when the protein is free in solution, both with and without membrane interactions. Furthermore, we found that αS adopts partially α-helical structure already after N-terminal tethering and upon adsorption to an unmodified gold surface. Hence, conformational changes of αS to α-helical structure can also be triggered by surface interactions in general. Our results are of importance for biotechnological applications and protein assays that rely on similar immobilization methods. A thorough understanding of immobilization effects on protein structure and function is crucial for the design of protein in vitro studies.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We gratefully acknowledge financial support by the Deutsche Forschungsgemeinschaft (SFB 969, A2 and SFB 1214, A3).
References
- V. M. Nemani, W. Lu, V. Berge, K. Nakamura, B. Onoa, M. K. Lee, F. A. Chaudhry, R. A. Nicoll and R. H. Edwards, Neuron, 2010, 65, 66–79 CrossRef CAS PubMed.
- P. K. Auluck, G. Caraveo and S. Lindquist, Annu. Rev. Cell Dev. Biol., 2010, 26, 211–233 CrossRef CAS PubMed.
- R. Bussell, T. F. Ramlall and D. Eliezer, Protein Sci., 2005, 14, 862–872 CrossRef CAS PubMed.
- M. Drescher, M. Huber and V. Subramaniam, ChemBioChem, 2012, 13, 761–768 CrossRef CAS PubMed.
- W. S. Davidson, A. Jonas, D. F. Clayton and J. M. George, J. Biol. Chem., 1998, 273, 9443–9449 CrossRef CAS PubMed.
- R. J. Perrin, W. S. Woods, D. F. Clayton and J. M. George, J. Biol. Chem., 2000, 275, 34393–34398 CrossRef CAS PubMed.
- M. Robotta, P. Braun, B. van Rooijen, V. Subramaniam, M. Huber and M. Drescher, ChemPhysChem, 2011, 12, 267–269 CrossRef CAS PubMed.
- I. Dikiy and D. Eliezer, Biochim. Biophys. Acta, Biomembr., 2012, 1818, 1013–1018 CrossRef CAS PubMed.
- M. A. Fallah, H. R. Gerding, C. Scheibe, M. Drescher, C. Karreman, S. Schildknecht, M. Leist and K. Hauser, ChemBioChem, 2017, 18, 2312–2316 CrossRef CAS PubMed.
- D. Eliezer, E. Kutluay, R. Bussell and G. Browne, J. Mol. Biol., 2001, 307, 1061–1073 CrossRef CAS PubMed.
- S. B. Lokappa and T. S. Ulmer, J. Biol. Chem., 2011, 286, 21450–21457 CrossRef CAS PubMed.
- M. Drescher, G. Veldhuis, B. D. van Rooijen, S. Milikisyants, V. Subramaniam and M. Huber, J. Am. Chem. Soc., 2008, 130, 7796–7797 CrossRef CAS PubMed.
- E. Jo, J. McLaurin, C. M. Yip, P. St George-Hyslop and P. E. Fraser, J. Biol. Chem., 2000, 275, 34328–34334 CrossRef CAS PubMed.
- L. Giehm, D. I. Svergun, D. E. Otzen and B. Vestergaard, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 3246–3251 CrossRef CAS PubMed.
- M. E. van Raaij, J. van Gestel, I. M. Segers-Nolten, S. W. de Leeuw and V. Subramaniam, Biophys. J., 2008, 95, 4871–4878 CrossRef CAS PubMed.
- R. Sarroukh, E. Goormaghtigh, J. M. Ruysschaert and V. Raussens, Biochim. Biophys. Acta, 2013, 1828, 2328–2338 CrossRef CAS PubMed.
- R. Mishra, B. Bulic, D. Sellin, S. Jha, H. Waldmann and R. Winter, Angew. Chem., Int. Ed., 2008, 47, 4679–4682 CrossRef CAS PubMed.
- G. Zandomeneghi, M. R. H. Krebs, M. G. Mccammon and M. Fandrich, Protein Sci., 2004, 13, 3314–3321 CrossRef CAS PubMed.
- H. Susi, Sn. Timashef and L. Stevens, J. Biol. Chem., 1967, 242, 5460–5466 CAS.
- M. S. Celej, R. Sarroukh, E. Goormaghtigh, G. D. Fidelio, J. M. Ruysschaert and V. Raussens, Biochem. J., 2012, 443, 719–726 CrossRef CAS PubMed.
- C. Kotting, J. Guldenhaupt and K. Gerwert, Chem. Phys., 2012, 396, 72–83 CrossRef.
- A. Nabers, J. Ollesch, J. Schartner, C. Kotting, J. Genius, U. Haussmann, H. Klafki, J. Wiltfang and K. Gerwert, J. Biophotonics, 2016, 9, 224–234 CrossRef CAS PubMed.
- A. Nabers, J. Ollesch, J. Schartner, C. Kotting, J. Genius, H. Hafermann, H. Klafki, K. Gerwert and J. Wiltfang, Anal. Chem., 2016, 88, 2755–2762 CrossRef CAS PubMed.
- R. F. Aroca, D. J. Ross and C. Domingo, Appl. Spectrosc., 2004, 58, 324a–338a CrossRef CAS PubMed.
- M. Osawa, Top. Appl. Phys., 2001, 81, 163–187 CrossRef CAS.
- G. Fusco, A. De Simone, T. Gopinath, V. Vostrikov, M. Vendruscolo, C. M. Dobson and G. Veglia, Nat. Commun., 2014, 5, 3827 CrossRef CAS PubMed.
- M. A. Fallah, C. Stanglmair, C. Pacholski and K. Hauser, Langmuir, 2016, 32, 7356–7364 CrossRef CAS PubMed.
- H. Miyake, S. Ye and M. Osawa, Electrochem. Commun., 2002, 4, 973–977 CrossRef CAS.
- K. Ataka and J. Heberle, Anal. Bioanal. Chem., 2007, 388, 47–54 CrossRef CAS PubMed.
- X. Jiang, E. Zaitseva, M. Schmidt, F. Siebert, M. Engelhard, R. Schlesinger, K. Ataka, R. Vogel and J. Heberle, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 12113–12117 CrossRef CAS PubMed.
- M. Mrksich and G. M. Whitesides, Annu. Rev. Biophys. Biomol. Struct., 1996, 25, 55–78 CrossRef CAS PubMed.
- Y. L. Wang, H. H. Lai, M. Bachman, C. E. Sims, G. P. Li and N. L. Allbritton, Anal. Chem., 2005, 77, 7539–7546 CrossRef CAS PubMed.
- S. I. Jeon, J. H. Lee, J. D. Andrade and P. G. Degennes, J. Colloid Interface Sci., 1991, 142, 149–158 CrossRef CAS.
- E. Ostuni, L. Yan and G. M. Whitesides, Colloids Surf., B, 1999, 15, 3–30 CrossRef CAS.
- M. T. L. Casford, A. Ge, P. J. N. Kett, S. Ye and P. B. Davies, J. Phys. Chem. B, 2014, 118, 3335–3345 CrossRef CAS PubMed.
- Y. Niu, A. I. Matos, L. M. Abrantes, A. S. Viana and G. Jin, Langmuir, 2012, 28, 17718–17725 CrossRef CAS PubMed.
- C. E. Jordan, B. L. Frey, S. Kornguth and R. M. Corn, Langmuir, 1994, 10, 3642–3648 CrossRef.
- T. Kang, S. Hong, H. J. Kim, J. Moon, S. Oh, S. R. Paik and J. Yi, Langmuir, 2006, 22, 13–17 CrossRef CAS PubMed.
- J. Schartner, J. Guldenhaupt, B. Mei, M. Rogner, M. Muhler, K. Gerwert and C. Kotting, J. Am. Chem. Soc., 2013, 135, 4079–4087 CrossRef CAS PubMed.
- K. Ataka and J. Heberle, Biopolymers, 2006, 82, 415–419 CrossRef CAS PubMed.
- B. L. Frey and R. M. Corn, Anal. Chem., 1996, 68, 3187–3193 CrossRef CAS.
- L. H. Dubois and R. G. Nuzzo, Annu. Rev. Phys. Chem., 1992, 43, 437–463 CrossRef CAS.
- P. Flagmeier, G. Meisl, M. Vendruscolo, T. P. J. Knowles, C. M. Dobson, A. K. Buell and C. Galvagnion, Proc. Natl. Acad.
Sci. U. S. A., 2016, 113, 10328–10333 CrossRef CAS PubMed.
- C. Galvagnion, A. K. Buell, G. Meisl, T. C. Michaels, M. Vendruscolo, T. P. Knowles and C. M. Dobson, Nat. Chem. Biol., 2015, 11, 229–234 CrossRef CAS PubMed.
- N. P. Reynolds, A. Soragni, M. Rabe, D. Verdes, E. Liverani, S. Handschin, R. Riek and S. Seeger, J. Am. Chem. Soc., 2011, 133, 19366–19375 CrossRef CAS PubMed.
- A. Iyer, N. O. Petersen, M. M. A. E. Claessens and V. Subramaniam, Biophys. J., 2014, 106, 2585–2594 CrossRef CAS PubMed.
- K. A. Conway, J. D. Harper and P. T. Lansbury Jr., Biochemistry, 2000, 39, 2552–2563 CrossRef CAS PubMed.
- C. Galvagnion, J. W. P. Brown, M. M. Ouberai, P. Flagmeier, M. Vendruscolo, A. K. Buell, E. Sparr and C. M. Dobson, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 7065–7070 CrossRef CAS PubMed.
- W. Hoyer, T. Antony, D. Cherny, G. Heim, T. M. Jovin and V. Subramaniam, J. Mol. Biol., 2002, 322, 383–393 CrossRef CAS PubMed.
- B. A. Silva, O. Einarsdottir, A. L. Fink and V. N. Uversky, Biomolecules, 2013, 3, 703–732 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9bm00140a |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*





