
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Micelle carriers based on dendritic macromolecules containing bis-MPA and glycine for antimalarial drug delivery†
Elisabet
Martí Coma-Cros‡
abc,
Alexandre
Lancelot‡
 d,
María
San Anselmo
d,
Livia
Neves Borgheti-Cardoso
abc,
Juan José
Valle-Delgado
e,
José Luis
Serrano
*df,
Xavier
Fernàndez-Busquets
*abc and
Teresa
Sierra
*f
d,
María
San Anselmo
d,
Livia
Neves Borgheti-Cardoso
abc,
Juan José
Valle-Delgado
e,
José Luis
Serrano
*df,
Xavier
Fernàndez-Busquets
*abc and
Teresa
Sierra
*f
aNanomalaria Group, Institute for Bioengineering of Catalonia (IBEC), The Barcelona Institute of Science and Technology, Baldiri Reixac 10-12, ES-08028 Barcelona, Spain. E-mail: xfernandez_busquets@ub.edu
bBarcelona Institute for Global Health (ISGlobal, Hospital Clínic-Universitat de Barcelona), Rosselló 149-153, ES-08036 Barcelona, Spain
cNanoscience and Nanotechnology Institute (IN2UB), University of Barcelona, Martí i Franquès 1, ES-08028 Barcelona, Spain
dInstituto de Nanociencia de Aragón (INA), Química Orgánica, Facultad de Ciencias, Universidad de Zaragoza, Spain. E-mail: joseluis@unizar.es
eDepartment of Bioproducts and Biosystems, School of Chemical Engineering, Aalto University, PO Box 16300, FI-00076, Aalto, Finland
fInstituto de Ciencia de Materiales de Aragón (ICMA), Facultad de Ciencias, Universidad de Zaragoza-CSIC, Spain. E-mail: tsierra@unizar.es
First published on 4th February 2019
Abstract
Biomaterials for antimalarial drug transport still need to be investigated in order to attain nanocarriers that can tackle essential issues related to malaria treatment, e.g. complying with size requirements and targeting specificity for their entry into Plasmodium-infected red blood cells (pRBCs), and limiting premature drug elimination or drug resistance evolution. Two types of dendritic macromolecule that can form vehicles suitable for antimalarial drug transport are herein explored. A new hybrid dendritic-linear-dendritic block copolymer based on Pluronic® F127 and amino terminated 2,2′-bis(glycyloxymethyl)propionic acid dendrons with a poly(ester amide) skeleton (HDLDBC-bGMPA) and an amino terminated dendronized hyperbranched polymer with a polyester skeleton derived from 2,2′-bis(hydroxymethyl)propionic acid (DHP-bMPA) have provided self-assembled and unimolecular micelles. Both types of micelle carrier are biocompatible and exhibit appropriate sizes to enter into pRBCs. Targeting studies have revealed different behaviors for each nanocarrier that may open new perspectives for antimalarial therapeutic approaches. Whereas DHP-bMPA exhibits a clear targeting specificity for pRBCs, HDLDBC-bGMPA is incorporated by all erythrocytes. It has also been observed that DHP-bMPA and HDLDBC-bGMPA incorporate into human umbilical vein endothelial cells with different subcellular localization, i.e. cytosolic and nuclear, respectively. Drug loading capacity and encapsulation efficiencies for the antimalarial compounds chloroquine, primaquine and quinacrine ranging from 30% to 60% have been determined for both carriers. The resulting drug-loaded nanocarriers have been tested for their capacity to inhibit Plasmodium growth in in vitro and in vivo assays.
Introduction
According to the latest estimates, around 219 million cases of malaria occurred globally in 2017 and the disease led to 435![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths.1 People living in the poorest countries are the most vulnerable, with approximately 90% of deaths in Africa, of which 70% are children under 5 years of age. Although increased prevention and control measures have led to a reduction in malaria mortality rates by more than 42% globally since 2000, an estimated 3.4 billion people are still at risk of being infected and developing disease. Despite the undeniable importance of malaria elimination on the global research agenda, current vaccines in development do not offer prospects of complete protection2 and the available front-line drugs are rapidly losing efficacy, with resistance already evolved to the first-line drug artemisinin.3 As a result, since 2014 the malaria incidence and mortality decline have stalled. Thus, alternative strategies4 working through radically new mechanisms are urgently needed. Antimalarial drugs can potentially target a suite of pathogen life stages inside two different hosts: humans and the insect vector. Infection starts when a parasitized female Anopheles mosquito, while taking a blood meal, inoculates sporozoites of the malaria parasite, the protist Plasmodium spp. In the liver, sporozoites develop into merozoites,5 which enter the circulation, invade red blood cells (RBCs)6 and replicate asexually through ring, trophozoite and schizont stages to produce daughter cells that invade new RBCs to perpetuate the blood-stage cycle. Some parasites eventually differentiate into sexual stages, female or male gametocytes that are ingested by a mosquito from peripheral blood. Following fertilization in the insect's midgut, the zygote differentiates into an ookinete that moves through the midgut epithelium and forms an oocyst, which releases sporozoites. The malaria transmission cycle is restarted when sporozoites migrate to the salivary glands and are injected into a human with the mosquito's next bite.
000 deaths.1 People living in the poorest countries are the most vulnerable, with approximately 90% of deaths in Africa, of which 70% are children under 5 years of age. Although increased prevention and control measures have led to a reduction in malaria mortality rates by more than 42% globally since 2000, an estimated 3.4 billion people are still at risk of being infected and developing disease. Despite the undeniable importance of malaria elimination on the global research agenda, current vaccines in development do not offer prospects of complete protection2 and the available front-line drugs are rapidly losing efficacy, with resistance already evolved to the first-line drug artemisinin.3 As a result, since 2014 the malaria incidence and mortality decline have stalled. Thus, alternative strategies4 working through radically new mechanisms are urgently needed. Antimalarial drugs can potentially target a suite of pathogen life stages inside two different hosts: humans and the insect vector. Infection starts when a parasitized female Anopheles mosquito, while taking a blood meal, inoculates sporozoites of the malaria parasite, the protist Plasmodium spp. In the liver, sporozoites develop into merozoites,5 which enter the circulation, invade red blood cells (RBCs)6 and replicate asexually through ring, trophozoite and schizont stages to produce daughter cells that invade new RBCs to perpetuate the blood-stage cycle. Some parasites eventually differentiate into sexual stages, female or male gametocytes that are ingested by a mosquito from peripheral blood. Following fertilization in the insect's midgut, the zygote differentiates into an ookinete that moves through the midgut epithelium and forms an oocyst, which releases sporozoites. The malaria transmission cycle is restarted when sporozoites migrate to the salivary glands and are injected into a human with the mosquito's next bite.
Most chemotherapeutic approaches against malaria are targeted at the asexual, blood-stage parasites that are responsible for all symptoms and pathologies of the disease.7 Currently administered antimalarial drugs are in free form in the blood circulation and have poor specificity for Plasmodium-infected RBCs (pRBCs). This incurs the risk of having to deliver large overall doses over an extended period of time to compensate for drug removal through spleen and liver clearance and kidney filtration. However, patient non-compliance and low concentration thresholds to ward off potential side effects often end up in sublethal local drug amounts reaching infected cells, which in turn stimulate resistance evolution. Because malaria pathophysiology is so complex and the disease is so widespread, it is generally accepted that to achieve eradication a combination of weapons will be needed.8 These include the improvement of existing approaches and the development of new ones,9 with drug therapy remaining the mainstay of treatment and prevention,10 and nanotechnology being able to provide innovative useful tools.11 The objective of delivering drugs exclusively to a selected site with minimal exposure for sensitive adjacent healthy cells or tissues is the holy grail of the fast-developing nanomedicine field.12 Encapsulation of drugs in targeted nanovectors is a rapidly growing area with a clear applicability to infectious disease treatment,13 and pharmaceutical nanotechnology has been identified as a potentially essential asset to the future fight against malaria.14,15
In the search for suitable nanocarriers for the treatment of infectious diseases, dendrimers stand out as valuable candidates due to their inherent features for building nanostructures with controlled size, morphology and surface functionalization.16 In a previous work we had explored different amphiphilic cationic dendritic derivatives based on 2,2′-bis(hydroxymethyl)propionic acid (bis-MPA)17 as nanocarriers for the targeted delivery of antimalarial drugs.18 These dendritic derivatives consisted of Janus dendrimers19 or hybrid dendritic-linear-dendritic block copolymers (HDLDBC),20 and both self-assembled into micelles with glycine groups at the surface. Although some of these structures exhibited specific pRBC targeting and in vitro antimalarial activity, they had an excessive unspecific toxicity, a very large size after antimalarial drug loading (>150 nm), and their in vivo tests had been preliminary.
Here we build on our previous work to explore new specifically targeted delivery systems for antimalarial drugs of a size after drug encapsulation (<30 nm) sufficiently small to facilitate their entry into pRBCs.21 Accordingly, two types of cationic dendritic derivative are proposed, which mainly differ in the way they form the nanocarrier, either self-assembled micelles or unimolecular micelles. As for the former, and on the basis of the promising results shown by the previously reported Pluronic® F127 HDLDBC derivative that contained polyester dendrons,18 we have herein conjugated Pluronic® F127 with poly(ester amide) dendrons to generate a new micelle-forming self-assembling amphiphilic cationic HDLDBC (Fig. 1A). The combination of the hydrolytic degradability of ester linkages and the stability and H-bond-forming ability of amide groups has proven very interesting for the design of synthetic polymers for biomedical applications.22,23 Accordingly, a poly(ester amide) dendron conjugated at both ends of Pluronic® F127 has been herein envisaged as a possibility to modulate the size and stability of self-assembled micelles given the additional H-bonding interactions due to amide groups. The dendron employed derives from the monomer 2,2′-bis(glycyloxymethyl)propionic acid (bis-GMPA), which was recently described by us as one of the few examples of poly(ester amide) dendritic structures reported in the literature, which showed high potential for biomedical applications due to the biocompatibility, degradability and positive results in drug and gene delivery of its dendritic derivatives.24 As for the achievement of a unimolecular micelle carrier, we have selected dendronized hyperbranched polymers (DHPs), also called pseudo-dendrimers, based on a hyperbranched polymer of bis-MPA, conjugated at its periphery with bis-MPA dendrons with exposed glycine moieties (Fig. 1B). We demonstrated that these DHP-bMPA derivatives could deliver DNA into mesenchymal cells.25 However, their potential as drug carriers remains to be explored. In this respect, these polymers showed appropriate biocompatibility and degradability that make them interesting to be investigated as carriers for antimalarial drug delivery.
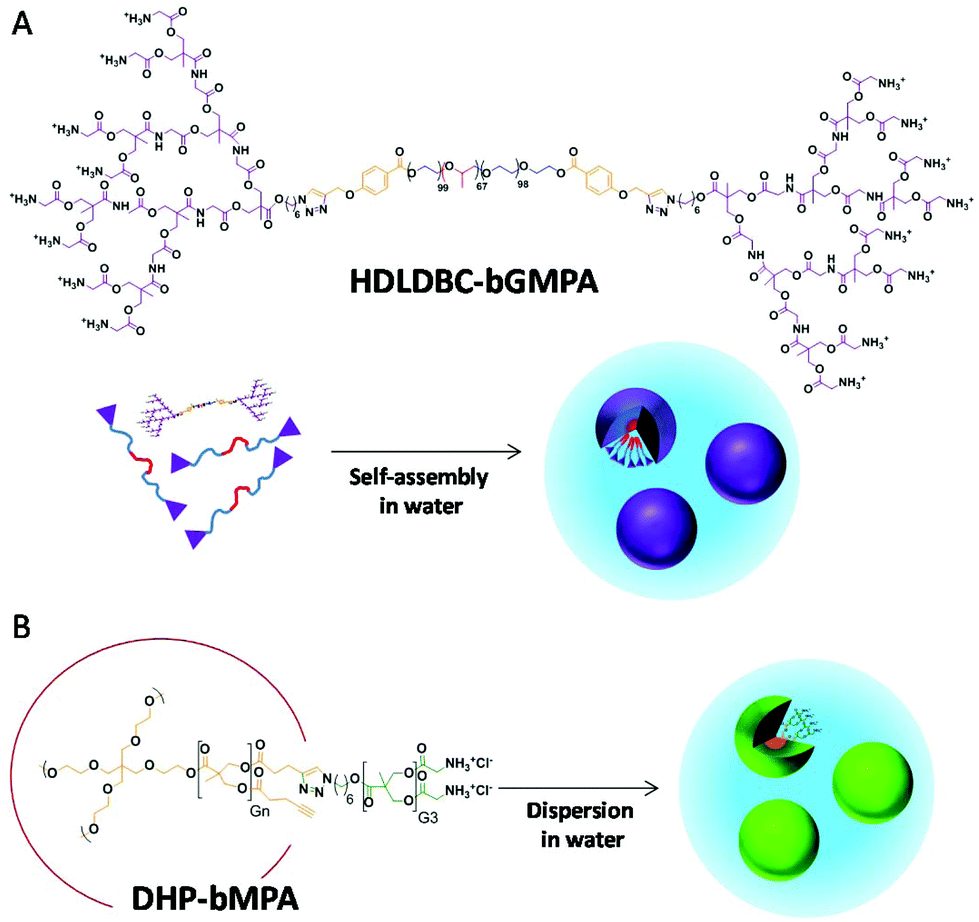 | ||
| Fig. 1 Chemical structure and cartoon representation of (A) micelle formation by HDLDBC-bGMPA, a hybrid dendritic-linear-dendritic block copolymer composed by Pluronic F127 and dendrons derived from 2,2′-bis(glycyloxymethyl)propionic acid (bis-GMPA), and (B) DHP-bMPA, dendronized hyperbranched polymers derived from 2,2′-bis(hydroxymethyl)propionic acid (bis-MPA) hyperbranched polymers and bis-MPA dendrons functionalized with glycine groups. | ||
Both types of structure have been investigated for their targeting towards pRBCs, and the drug loading capacity and encapsulation efficiencies of the carriers have been tested with the antimalarial compounds chloroquine (CQ), primaquine (PQ) and quinacrine (QN). The resulting nanocarriers have been assayed for their capacity to inhibit Plasmodium growth in in vitro and in vivo assays.
Materials and methods
Reagents
Unless otherwise indicated, all reagents were purchased from Sigma-Aldrich® or Acros™, and used always without further purification. Dichloromethane (DCM) and tetrahydrofuran (THF) were dried using solvent purification systems.Synthesis and characterization of the dendritic derivatives
Globular DHP-bMPA dendronized hyperbranched polymers as well as DHP-bMPA-Rho, a DHP containing covalently linked rhodamine B fluorophore, were synthesized as previously reported by us.25 HDLDBC-bGMPA was prepared by copper azide–alkyne cycloaddition (CuAAC) coupling of the t-Boc protected bis-GMPA dendron and the Pluronic® F127 bis(alkyne) derivative as depicted in Fig. 2A.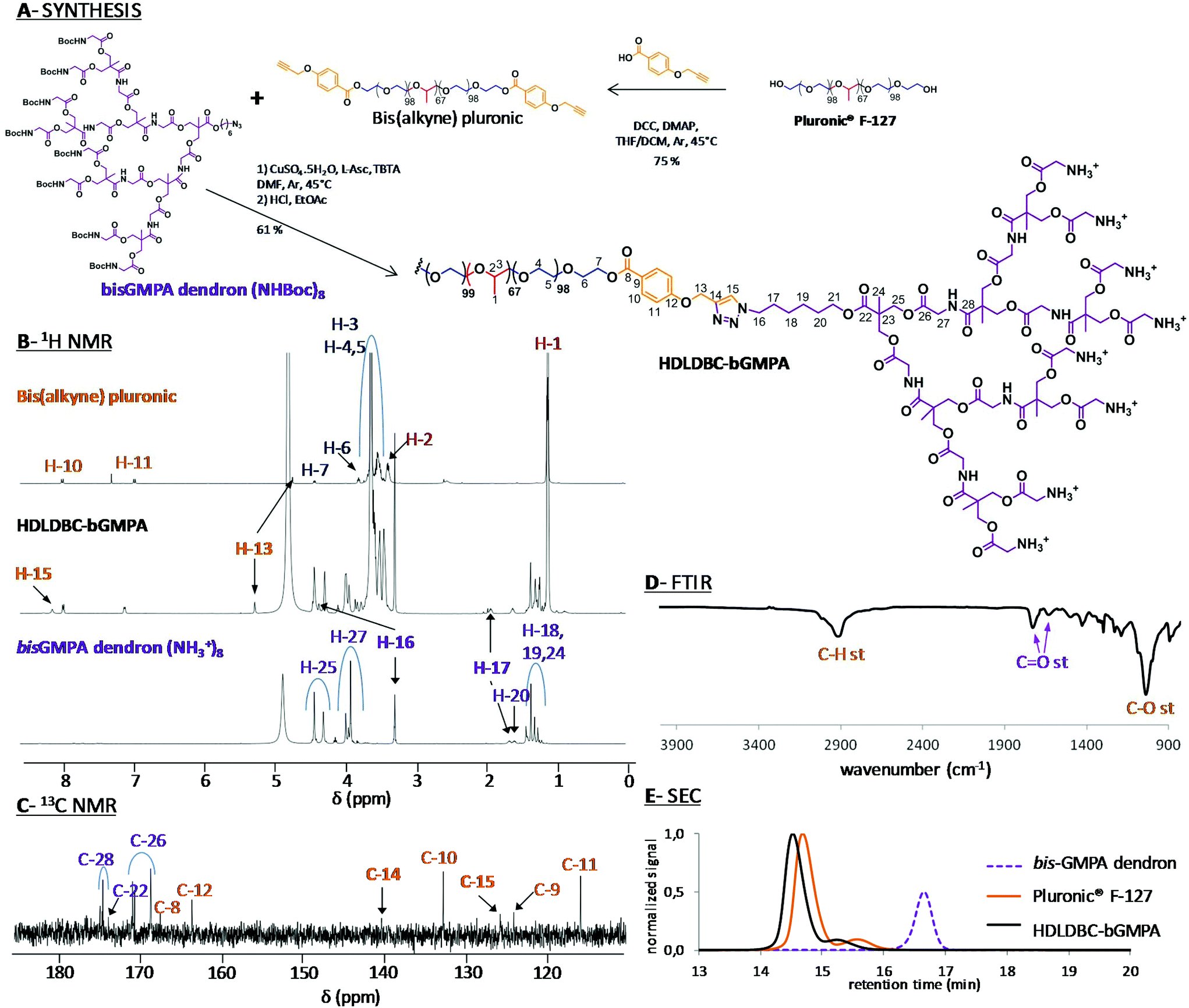 | ||
| Fig. 2 Synthesis of HDLDBC-bGMPA (A) and chemical characterization by 1H NMR (B), 13C NMR (C), FTIR (D) and SEC (E). 1H NMR spectra with signal relative integration, the 1H–1H COSY NMR spectra and full 13C NMR spectra of HDLDBC-bGMPA are gathered in the ESI.† | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O st ester), 1718 (CO st carbamate), 1670 (CO st amide), 1533 (N–H δ), 1468 (CH2, –CH3δ), 1101 (C–O–C st). SEC (ref PMMA): 2 populations; Mw 19745 g mol−1; Đ: 1.08.
O st ester), 1663 (CO st amide and N–H+δ), 1545 (N–H δ), 1468 (CH2–, CH3δ), 1099 (C–O–C st).
O st ester), 1718 (CO st carbamate), 1670 (CO st amide), 1533 (N–H δ), 1468 (CH2, –CH3δ), 1101 (C–O–C st). SEC (ref PMMA): 2 populations; Mw 19745 g mol−1; Đ: 1.08.
O st ester), 1663 (CO st amide and N–H+δ), 1545 (N–H δ), 1468 (CH2–, CH3δ), 1099 (C–O–C st).
Self-assembly of HDLDBC-bGMPA
HDLDBC-bGMPA was mixed with the corresponding amount of distilled water and cooled down to 4 °C for 30 min until complete dissolution. Then, the solution was slowly heated to room temperature to trigger the formation of the water soluble nanocarriers. The critical micelle concentration (CMC) was determined using the Nile Red technique.26 Briefly, 2 mL of distilled water solutions of HDLDBC-bGMPA with concentrations ranging from 0.1 to 5 mg mL−1 were prepared at 25 °C. Nile Red was dissolved in ethanol at a concentration of 0.25 mM and 10 μL of this solution was added to each sample. The mixtures were then stirred at room temperature for 1 h in the dark with an orbital shaker. The fluorescence emission spectrum of each solution was recorded with a PerkinElmer LS 55 fluorimeter after excitation at λex = 550 nm.Preparation of carrier-antimalarial drug conjugates and drug release assays
The antimalarial compounds CQ, PQ and QN were encapsulated within the corresponding carrier (DHP-bMPA or HDLDBC-bGMPA) following the oil-in-water procedure previously employed by us for CQ and PQ.18 Briefly, each dendritic derivative and the corresponding drug were dissolved into a mixture of DCM and distilled water (1:1) at feeding ratios of wDHP:wdrug = 1:0.5 and wHDLDBC:wdrug = 1:1. The samples were vigorously stirred at room temperature employing an orbital shaker under ventilation until complete evaporation of DCM (around 2 h). Non-encapsulated drug was removed by dialysis (regenerated cellulose membrane, MW 1000 Da cut off, Spectra/Por®) against distilled water (200 mL) at 4 °C for 16 h. The amount of encapsulated drug was indirectly determined: the quantity of drug present in the dialysis water was measured by UV-VIS spectrometry (Varian Cary50 Probe UV-visible spectrophotometer) at the wavelengths of λA(CQ) = 345 nm for CQ, λA(PQ) = 259 nm for PQ and λA(QN) = 280 nm for QN and was subtracted from the initially incorporated drug. The samples were freeze-dried with a Telstar Cryodos 50 freeze-dryer in order to increase their stability over long storage periods.
The release of CQ encapsulated within HDLDBC-bGMPA and DHP-bMPA was studied by dialysis against phosphate buffered saline (PBS). 2 mL of the dendrimer/drug conjugates were dialyzed against 200 mL of PBS (regenerated cellulose dialysis membrane, MW 1000 Da cut-off, Spectra/Por®) under stirring at 37 °C. For CQ determination, aliquots (2 mL) were withdrawn at different times from the waters of dialysis up to 72 h (specifically at 0, 2, 4, 6, 24, 48, and 72 h). An identical procedure was performed with a control solution of free CQ at the same concentration as in the conjugate preparation.
Fluorescent labeling of HDLDBC-bGMPA
HDLDBC-bGMPA nanocarriers were labeled by encapsulating a low water soluble modified rhodamine B (Rho(C17)2) red fluorophore previously reported by us,27 and the oil-in-water procedure was employed to encapsulate the fluorophore within the nanocarrier. The selected feeding ratio in this case was (1:0.15) (wHDLDBC:wRho). The release profile of Rho(C17)2 from the dendrimer/Rho(C17)2 conjugates was studied by dialysis similarly as the drug release procedure described above. In this case, 2 mL of the HDLDBC-bGMPA/Rho(C17)2 conjugate containing 0.15 mg mL−1 of encapsulated Rho(C17)2 were dialyzed (regenerated cellulose membrane, MW 2000 Da cut off, Spectra/Por®) against distilled water (200 mL) at 37 °C. At different times up to 72 h, 2 mL aliquots were withdrawn from where the quantity of Rho(C17)2 was determined by measuring fluorescence intensity (λex = 540 nm, λem = 580 nm).
Transmission electron microscopy (TEM) and atomic force microscopy (AFM) analysis
TEM images were obtained with a FEI TECNAI T20 electron microscope (FEI Company, Eindhoven, The Netherlands) with 200 kV beam power, using holey carbon film 300 mesh coppered grids (Agar Scientific Ltd). A droplet of an aqueous solution of the sample at a concentration of 1 mg mL−1 was deposited on the grid and let to adsorb for 30 s, removing the excess aqueous solution by blotting with filter paper. A droplet of a 3% w/v aqueous solution of phosphotungstic acid used as negative stain was deposited on the grid and left for 10 s before removing the excess staining solution by blotting with filter paper. The grid was dried for at least 24 h under atmospheric pressure at room temperature. The average size of the different nanocarriers was obtained by analysing ≥100 structures in ≥4 TEM images.For AFM analyses, 10 μL of 1 μg mL−1 or 10 ng mL−1 dendrimer solution in double deionised water (ddH2O; MilliQ system, Millipore) were deposited on cleaved mica substrates and, after an adsorption time of about 5 min, 40 μL ddH2O were added. High-resolution images were obtained with a MultiMode 8 atomic force microscope equipped with a NanoScope V controller (Bruker Corporation) operating in ScanAsyst mode in liquid, using ScanAsyst-Fluid + probes (Bruker Corporation).
Plasmodium falciparum cell culture and parasite growth inhibition assay
P. falciparum 3D7 was grown in vitro in human RBCs of blood group type B prepared as described elsewhere28 using previously established conditions.29 Briefly, parasites (thawed from glycerol stocks) were cultured at 37 °C in T25 flasks (SPL Life Sciences) containing RBCs in Roswell Park Memorial Institute (RPMI) complete medium (supplemented with 5 g L−1 Albumax II and 2 mM glutamine) under a gas mixture of 92% N2, 5% CO2, and 3% O2. Synchronized ring stage cultures were obtained by 5% sorbitol lysis and synchronized late stage cultures were obtained using 70% Percoll® (GE Healthcare) purification to enrich in late trophozoites and early schizonts;30 the medium was changed every 2 days maintaining 3% hematocrit. For culture maintenance, parasitemias were kept below 5% late forms by dilution with fresh RBCs. The human blood used in this work was commercially obtained from the Banc de Sang i Teixits (http://www.bancsang.net). Blood was not specifically collected for this research; the purchased units had been discarded for transfusion, usually because of an excess of blood relative to anticoagulant solution. Prior to their use, blood units underwent the analytical checks specified in the current legislation. Before being delivered to us, unit data were anonymized and irreversibly dissociated, and any identification tag or label had been removed in order to guarantee the non-identification of the blood donor. No blood data were or will be supplied, in accordance with the current Ley Orgánica de Protección de Datos and Ley de Investigación Biomédica. The blood samples will not be used for studies other than those made explicit in this research. Experiments were approved by the Ethics Committee of the Hospital Clínic de Barcelona.To isolate P. falciparum merozoites, a 3D7 strain culture was tightly synchronized (sorbitol lysis on day 1, 70% Percoll® followed after 2 h by sorbitol on day 4, sorbitol on day 6), and after a further 40 h a final 70% Percoll® treatment was done. Purified late stages were cultured in 10 mL of complete RPMI without adding fresh RBCs, and when the majority of parasites were segmented schizonts, E-64 protease inhibitor was added to a final concentration of 10 μM. Between 6 to 10 h later, the culture was centrifuged at 730g for 8 min and the pelleted cells were taken up in 20 mL of PBS. The suspension was passed sequentially through a 18G needle and a 1.2 μm filter (Sartorius Stedim® Minisart) blocked for 30 min with PBS/1% BSA. Merozoites in PBS were collected in a 50 mL Falcon tube previously blocked for 1 h with PBS/1% BSA, and finally centrifuged at 2200g for 10 min. The supernatant was removed leaving about 200 μL of a merozoite suspension in PBS, which was immediately used for targeting analysis.
For growth inhibition assays, P. falciparum 3D7 cultures were adjusted to 3% hematocrit and 1.5% parasitemia with more than 90% of parasites at ring stage after sorbitol synchronization. 75 μL of these Plasmodium cultures were plated in 96-well plates and incubated for 40 h at 37 °C in the presence of free drugs and dendrimer–drug conjugates (added in a volume of 75 μL). After addition of 0.5 μM Syto-11 (Thermo Fisher Scientific, Inc.), parasitemia was determined by flow cytometry as previously described.28
Cytotoxicity and hemolysis assays
Human umbilical vein endothelial cells (HUVEC, ATCC) were cultured in Medium 199 (M199, LabClinics) supplemented with 10% heat-inactivated foetal bovine serum (FBS, PAA Laboratories, Germany), 1% each penicillin/streptomycin (Biological Industries), and 10 mM glutamine (complete M199). 5000 cells per well were plated in 96-well plates (Thermo Fisher Scientific Inc.) and after 24 h at 37 °C in 5% CO2 atmosphere the medium was substituted by dilutions of the dendrimers in 100 μL of culture medium without FBS, and incubation was resumed for 48 h. 10 μL of 4-[3-(4-iodophenyl)-2-(4-nitrophenyl)-2H-5-tetrazolio]-1,3-benzene disulfonate labeling reagent (WST-1, Roche Diagnostics GmbH) was added to each well, and the plate was incubated in the same conditions for a further 3 h. After thoroughly mixing for 1 min on a shaker, the absorbance of the samples was measured at 440 nm using a Benchmark Plus microplate reader (Bio-Rad Laboratories Inc.). WST-1 in the absence of cells was used as blank and samples were prepared in triplicate for each experiment, including positive (10% bleach) and negative (PBS) controls.For hemolysis assays, human blood collected in citrate-phosphate-dextrose buffer was washed as described previously.28 After the last washing step RBCs were diluted in PBS to yield a solution with 6% hematrocrit. 100 μL of RBCs from this suspension and 100 μL of a dendrimer sample were added to a 96-well plate. Each assay was performed in triplicate, including positive (1% Triton X-100) and negative (PBS) controls. After incubating for 3 h at 37 °C in 90% N2, 5% CO2, and 5% O2, samples were collected in eppendorf tubes, spun at 16000g for 5 min, and the supernatant absorbance was finally measured at 541 nm.
Cell targeting analysis
400 μL of desynchronized living P. falciparum 3D7 cultures were stained for 30 min with 4 μg mL−1 of the DNA dye Hoechst 33342. HUVEC (1.5 × 104 cells per cm2) were treated equally but for the use of complete M199 as culture medium, and nuclei staining done at day 3 after plating (10 μg Hoechst 33342 per mL). After 3 washes with complete RPMI or M199, respectively, Plasmodium cultures and HUVEC were incubated in the presence of 0.15 mg mL−1 dendrimer conjugated to rhodamine, for 90 min in the corresponding medium at 37 °C with gentle stirring. Purified merozoites were incubated for 10 min at room temperature (RT) in the simultaneous presence of Hoechst 33342 and rhodamine-labeled dendrimers. After washing with incomplete medium, nonfixed samples were placed in a 8-well LabTek chamber slide system (Lab-Tek®II, catalog number 155409) and the fluorescence of Hoechst 33342, FITC and rhodamine (λex/em: 350/461, 488/520 and 553/627 nm, respectively) was observed with an IX51 inverted fluorescence microscope (Olympus). Confocal fluorescence microscopy analysis was done with a Leica TCS SP5 laser scanning confocal microscope equipped with a DMI6000 inverted microscope, blue diode (405 nm), Argon (458/476/488/496/514 nm), diode pumped solid state (561 nm) and Helium–Neon (594/633 nm) lasers and PLAN APO 63× oil (NA 1.4) immersion objective lens. For flow cytometry analysis, Plasmodium cultures were diluted in PBS to a final concentration of 1–10 × 106 cells per mL, and samples were analyzed using a LSRFortessa™ flow cytometer instrument (BD Biosciences) set up with the 5 lasers, 20 parameters standard configuration. The single-cell population was selected on a forward-side scatter scattergram. Rhodamine was excited using a yellow-green laser (561 nm), and its fluorescence collected through a 582/15 nm filter. Hoechst 33342 was excited with a violet laser (405 nm), and its fluorescence collected using a 450/40 nm filter.Determination of the maximum tolerated dose (MTD) in mice
Seven week-old inbred BALB/cAnR mouse females (18–20 g, Janvier Laboratories) were maintained under standard environmental conditions (20–24 °C and 12 h/12 h light/dark cycle) with ad libitum access to food and water. To ensure administration and minimize injection stress, the animals were anesthetized with isoflurane (4% for induction and 2.5% for maintenance) in an oxygen stream, while a 250 μL bolus was administered intravenously. To reduce the number of animals used, an adaptation of OECD 425 Test Guideline was followed, which consisted of a single ordered dose progression. The first mouse received the dose initially planned for antimalarial activity assays (see below), and the dose for the next animal was either increased or decreased by a factor of 3.2 depending on whether the first animal survived or died, respectively. Each mouse was injected and evaluated for at least 48 h before the next animal was treated. All animals were observed for toxic signs during 14 days after dose injection. Following this protocol, three different concentrations of the dendrimers (11.7, 37.5 and 120 mg kg−1) were evaluated, prepared in PBS from a 50 mg mL−1 stock solution of dendrimers in sterile ddH2O. In the presence of toxic effects including, among others, >20% reduction in animal weight, aggressive and unexpected animal behavior or the presence of blood in faeces, animals were immediately anesthetized using a 100 mg kg−1 Ketolar plus 5 mg kg−1 Midazolan mixture and sacrificed by cervical dislocation. Dendrimer MTD was therefore defined upon completion of the assay as the highest dosage exhibiting an absence of the aforesaid toxicity signs. The animal care and use protocols followed adhered to the specific national and international guidelines specified in the Spanish Royal Decree 53/2013, which is based on the European regulation 2010/63/UE. The studies reported here were performed under protocols reviewed and approved by the Ethical Committee on Clinical Research from the Hospital Clínic de Barcelona (Reg. HCB/2014/0910).Antimalarial activity assay in vivo and dot-blot assays
The in vivo antimalarial activity of free CQ and of dendrimer-CQ conjugates was analyzed in a 4-day blood suppressive test as previously described.31 Briefly, BALB/c mice were inoculated intraperitoneally with 2 × 106 RBCs from Plasmodium yoelii yoelii 17XL (PyL) MRA-267-infected mice. Treatment started 4 h later (day 0) with a single dose of 1.9 mg CQ per kg per day administered intravenously as diphosphate–drug or dendrimer–drug, followed by identical dose administration for the next 3 days. Tested compounds were prepared in PBS and the control groups received PBS. Parasitemia was monitored daily by microscopic examination of Giemsa-stained thin blood smears using the Plasmoscore 1.3 software (Burnet Institute, Melbourne, Australia). When all surviving animals had completely cleared Plasmodium infection they were re-infected (on day 69) with P. yoelii yoelii 17XL (PyL) MRA-267 as above and left untreated. Survival was monitored until day 90, when blood samples (400 μL) were collected on 10 μL of 10% EDTA, centrifuged for 5 min (470g), and the obtained plasma supernatants were stored at −20 °C. 3 μL of P. yoelii yoelii 17XL (PyL) MRA-267 extracts32 containing 1.5 μg protein were applied and allowed to dry on a nitrocellulose membrane (0.45 μm, Bio-Rad, catalog number 1620145). Non-specific sites were blocked with 5% BSA in TBS-T (0.05% Tween 20, 150 mM NaCl, 20 mM Tris-HCl, pH 7.5) for 1 h at RT. Then the membrane was first incubated (30 min, RT) with the plasma supernatants dissolved in TBS-T (1:5000 dilution), washed (TBS-T, 3 × 5 min), and then treated with a secondary goat anti-mouse antibody conjugated to horseradish peroxidase (Millipore) in TBS-T (1:10000 dilution; 30 min, RT), followed by 3 × 5 min washes with TBS-T and one wash with TBS (TBS-T without Tween). Finally the membrane was incubated with ECL Prime Western Blotting Detection Reagent (Luminol, Amersham) for 30–60 s and scanned (ImageQuant LAS4000, GE Healthcare).
Statistical analysis
Data are presented as the mean ± standard error of at least three independent experiments, and the corresponding standard errors in histograms and graphs are represented by error bars. Percentages of viability were obtained using non-treated cells as control of survival and IC50 values were calculated by nonlinear regression with an inhibitory dose–response model using GraphPad Prism5 software. Concentrations were transformed using natural log for linear regression. Regression models were adjusted for replicates and assay data.Results and discussion
Preparation and characterization of the nanocarriers
The correct grafting of the two bis-GMPA dendrons at the extremities of Pluronic® F-127 was first controlled by 1H (Fig. 2B) and 13C (Fig. 2C) NMR experiments (see Fig. S1.1, S1.2 and S1.3† for 1H–1H COSY and full 1H and 13C NMR spectra). The appearance of a peak at 8.19 ppm (H-15) in the 1H NMR spectrum and two peaks at 125.8 (C-15) and 140.4 ppm (C-14) in the 13C NMR spectrum confirmed the formation of the triazole rings. Additionally, three other signals were shifted downfield in the 1H NMR spectra of the final HDLDBC when compared with the spectra of its two precursors. Thus, the signal corresponding to the methylene protons in the α-position of the triazole rings belonging to the linear polymeric part (H-13) was shifted from 4.72 to 5.30 ppm. The signals corresponding to the methylene protons in the α- and β-positions of the triazole rings belonging to the dendritic part (H-16 and H-17) were shifted from 3.20 and 1.65 to 4.45 and 1.95 ppm, respectively. Moreover, the relative integrations of the signals corresponding to the dendrons and linear polymer corroborated the grafting of two dendrons at the terminal position of the Pluronic® F-127 (Table S1.1†).
The characteristic bands of the bis-GMPA dendrons and Pluronic® F-127 could be observed in the FTIR spectrum, (Fig. 2D). First, the presence of the poly(ether) Pluronic® F-127 was asserted by the intense band at 1101 cm−1 (C–O bond vibration stretching) and the two intense bands at 2881 and 1344 cm−1 (C–H bond vibrations). Second, the presence of the poly(ester amide) bis-GMPA dendrons was confirmed by the two bands at 1755 and 1664 cm−1 corresponding to CO bond vibration stretching of ester and amide groups, respectively.
SEC was performed with the t-Boc-protected precursor of HDLDBC-bGMPA due to its low solubility in the elution solvent employed for chromatography, i.e. THF (Fig. 2E). Two peaks were observed in the chromatograms of both, the commercial Pluronic® F-127 and the HDLDBC, showing the polydispersity of these compounds. The correct functionalization of Pluronic® was nevertheless asserted as both peaks corresponding to it showed lower retention time than the two peaks corresponding to the starting Pluronic® F-127. Additionally, no peak corresponding to residual free bis-GMPA dendron could be observed. In summary, all experimental data obtained during the characterization of HDLDBC-bGMPA confirmed the correct insertion of two bis-GMPA dendrons at each extremity of the linear polymer and the absence of unreacted free linear polymer or dendron.
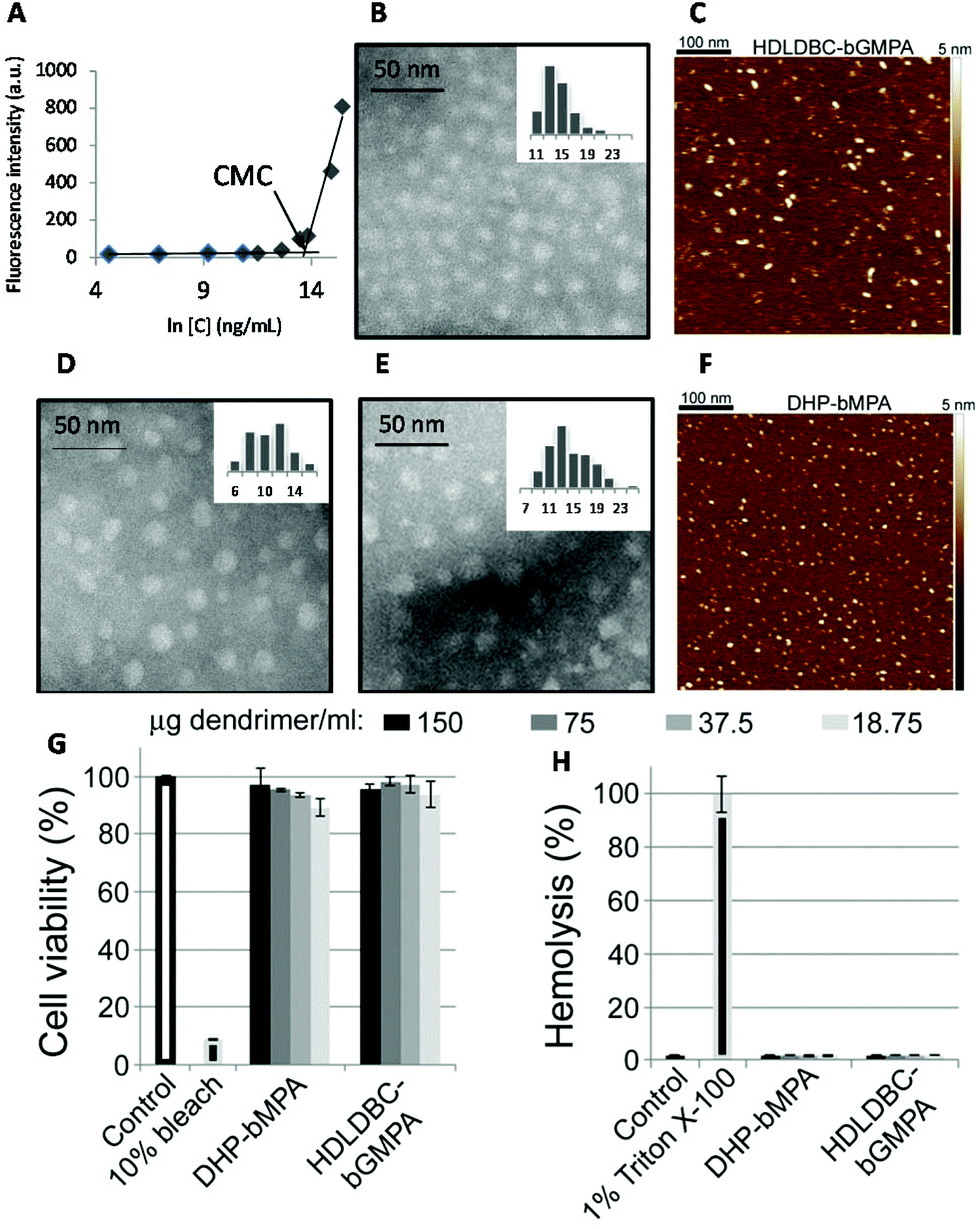 | ||
| Fig. 3 Characterization of nanocarriers. Nile Red fluorescence intensity as a function of HDLDBC-bGMPA concentration (A), TEM image (B) and AFM image in water on mica surface (C) of HDLDBC-bGMPA, TEM images of DHP-bMPA (n = 3 and n = 4 in Fig. 1B; D and E, respectively), and AFM image of DHP-bMPA (n = 4 in Fig. 1B) in water on mica surface (F). Cytotoxicity (G) and hemolysis assay (H) of the largest DHP-bMPA (n = 4 in Fig. 1B) and HDLDBC-bGMPA. | ||
The morphology of two globular dendronized hyperbranched polymers, DHP-bMPA, was also analyzed by TEM and AFM (Fig. 3D–F). The two generations tested (n = 3 and n = 4 in Fig. 1B) appeared as rounded objects, with average diameters calculated from TEM images of 9.8 ± 2.7 nm and 13.5 ± 3.5 nm, respectively. These dimensions are consistent with the expected formation by these dendronized hyperbranched polymers of unimolecular micelles well dispersed in aqueous solution.33,34 DLS measurements of DHP-bMPA (n = 4) gave a diameter of 11 ± 2 nm (Fig. S S2.1.B†). As for HDLDBC-bGMPA carriers, the size of DHP-bMPA unimolecular micelles results a priori appropriate to enter into pRBCs.21
In vitro targeting analysis
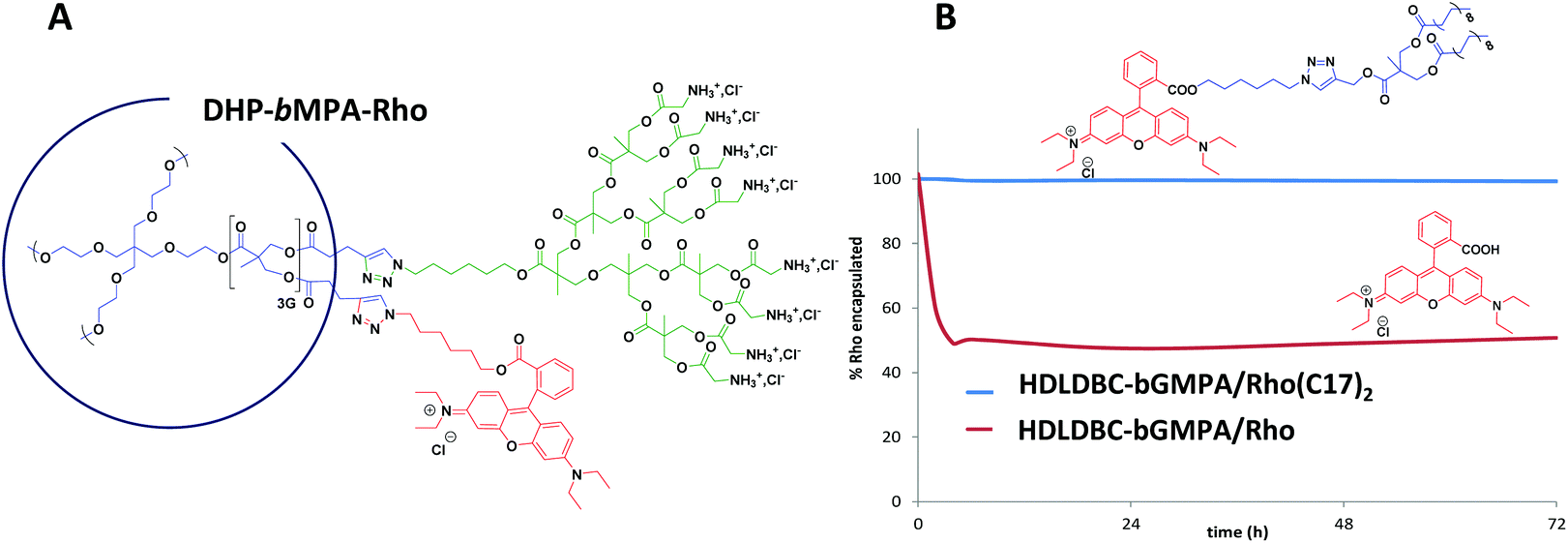 | ||
| Fig. 4 Rhodamine labeling of the nanocarriers. Chemical structure of the covalently labeled DHP-bMPA-Rho derivative (A), and release profile of the fluorescent rhodamine labels, Rho(C17)2 and Rho, from the nanocarrier HDLDBC-bGMPA (B). | ||
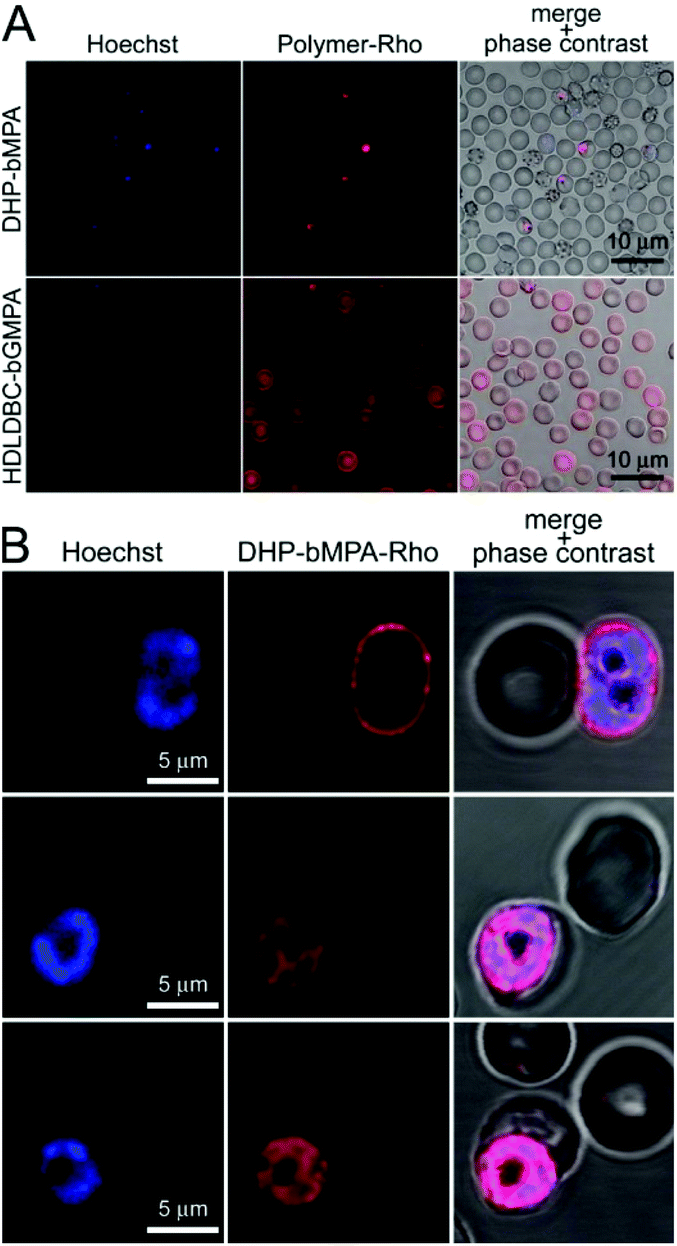 | ||
| Fig. 5 Fluorescence microscopy cell targeting analysis of rhodamine-labeled DHP-bMPA and HDLDBC-bGMPA to non-fixed RBCs and pRBCs. (A) Conventional fluorescence microscopy cell targeting of both polymers. (B) Confocal fluorescence microscopy cellular and subcellular targeting of DHP-bMPA-Rho. | ||
The differential RBC targeting of DHP-bMPA and HDLDBC-bGMPA was confirmed by flow cytometry assays (Fig. 6); whereas HDLDBC-bGMPA associated with P. falciparum merozoites and with both parasitized and non-parasitized erythrocytes, DHP-bMPA interacted only with merozoites and with pRBCs. HDLDBC-bGMPA interaction with both RBCs and pRBCs does not invalidate it as a potential carrier of antimalarial drugs in targeted delivery strategies. As we have shown before, delivering antimalarial compounds to non-infected erythrocytes might represent an interesting therapeutic approach whereby Plasmodium would encounter a hostile environment since the very first moment after RBC invasion.35,36 This scenario resulted in a significantly improved efficacy of drugs encapsulated inside liposomes targeted to both pRBCs and to non-parasitized red blood cells.35,37
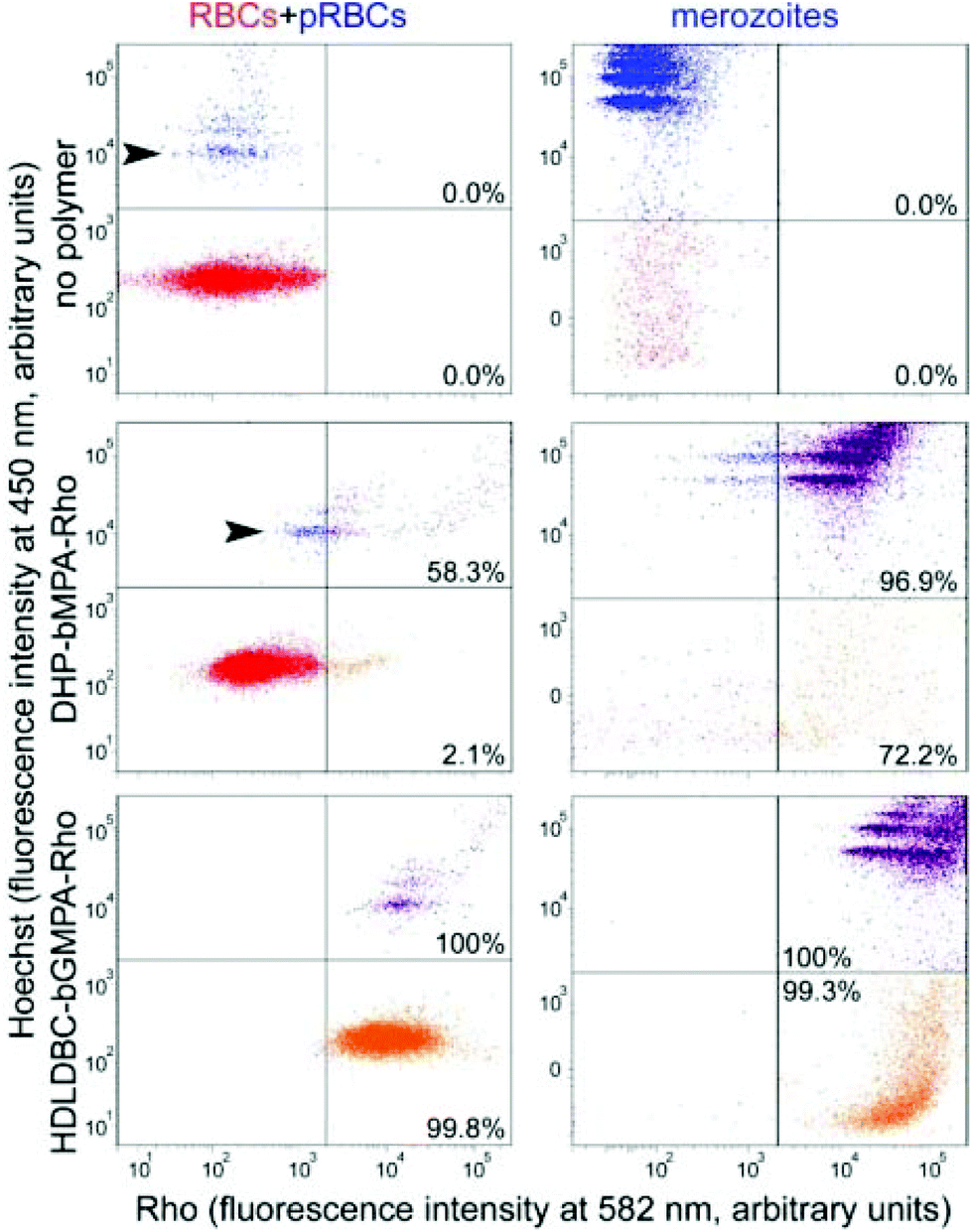 | ||
| Fig. 6 Flow cytometry analysis of the targeting of rhodamine-labeled DHP-bMPA and HDLDBC-bGMPA to RBCs, pRBCs and merozoites (the latter two represented in blue, upper left areas; their DNA is stained by Hoechst 33342). pRBCs and merozoites positive for rhodamine are shown in purple (upper right areas) whereas orange (lower right areas) indicates cells or cell debris (the latter only in merozoite samples) lacking DNA that are associated to the polymers. Percentages indicate the fraction of rhodamine-positive counts relative to the total number of recorded cells or particles in each sample. Arrowheads indicate ring stages. | ||
The modest binding of DHP-bMPA to early ring forms, where the parasite has not significantly modified the erythrocyte membrane, suggests that this polymer might interact predominantly with exported Plasmodium antigens, which are scarce in rings and completely absent from non-parasitized RBCs.
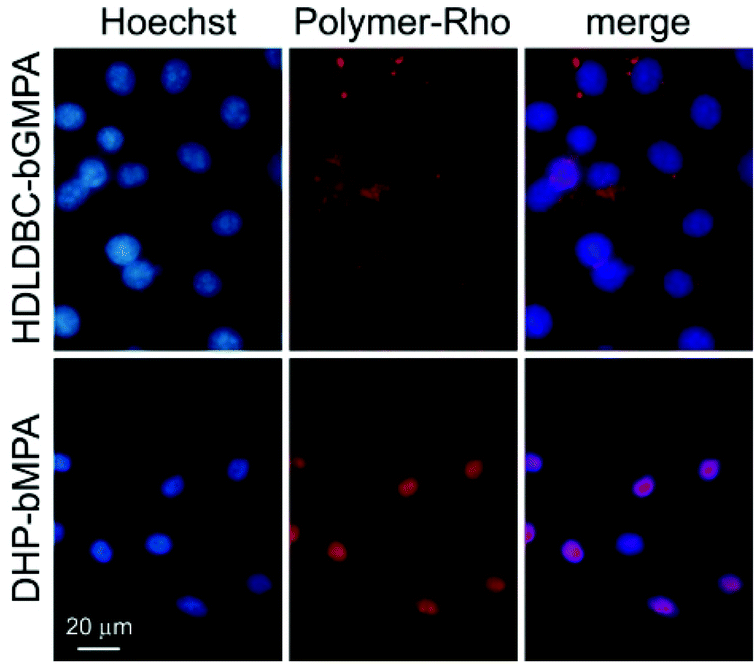 | ||
| Fig. 7 Fluorescence microscopy cell targeting analysis of rhodamine-labeled DHP-bMPA and HDLDBC-bGMPA to HUVEC cells. | ||
In vitro antimalarial activity of drug-loaded nanocarriers
:0.5 (wDHP/wdrug) and 1:1 (wHDLDBC/wdrug), respectively. This procedure allowed to encapsulate all three antimalarial drugs in DHP-bMPA nanocarriers with drug loading contents around 20% in weight and good encapsulation efficiencies, ranging from 37% to 60% (Table 1). A smaller DHP-bMPA (n = 3 in Fig. 1B) was also used to encapsulate all three antimalarials, obtaining similar drug contents and loading efficiencies (Table S2.1†). As for the HDLDBC-bGMPA carrier, higher loading capacities, between 30% and 48% in weight, and rather good encapsulation efficiencies, ranging from 31% to 48%, were attained.
| Drug | mg drug/mg carrier | EEa (%) | Average diameter (nm) | |
|---|---|---|---|---|
| a Encapsulation efficiency (EE, %) represents the fraction of encapsulated drug relative to the quantity used to perform the encapsulation. | ||||
| Empty | 13 ± 3 | |||
| HDLDBC-bGMPA | CQ | 0.307 | 31 | 17 ± 3 |
| PQ | 0.408 | 41 | 11 ± 2 | |
| QN | 0.475 | 48 | 14 ± 6 | |
| Empty | 13.5 ± 3.5 | |||
| DHP-bMPA | CQ | 0.246 | 60 | 20 ± 7 |
| PQ | 0.214 | 60 | 19 ± 8 | |
| QN | 0.229 | 37 | 12 ± 3 | |
All the aqueous solutions obtained after the encapsulation procedure were freeze-dried in order to enhance stability during storage of the carrier/drug systems. The effective encapsulation of drugs was asserted by comparing the absorption spectra of the drugs in water with those of the re-dissolved carrier/drug freeze-dried conjugates (Fig. S2.3 and S2.4†). In all cases, the differences observed reflect interactions between nanocarriers and drugs.
TEM images of drug-containing nanocarriers showed in all cases homogeneous dispersions of rounded objects (Fig. 8), the diameters of which were similar to those of empty nanocarriers, and always ≤20 nm (Table 1). This small size should favor their entry in pRBCs, since Plasmodium induces new permeation pathways that confer to the host cell an increased permeability to a wide range of particles up to diameters of 50–70 nm.21 Whereas the unimolecular micelles formed by DHP-bMPA in water (Fig. 8A) had an average diameter around 13.5 nm, DHP-bMPA/drug systems had an average diameter between 12 and 20 nm. Likewise, the loading of antimalarial drugs within HDLDBC-bGMPA nanocarriers resulted in structures with average sizes ranging from 11 to 17 nm (Fig. 8B), whereas the empty nanocarrier had an average size around 13 nm. AFM observation of both CQ-loaded carriers confirmed the formation of these rounded objects (Fig. 8).
 | ||
| Fig. 8 Negatively stained TEM images of the drug-loaded nanocarriers HDLDBC-bGMPA (A) and DHP-bMPA (B). Insets show AFM images of the corresponding CQ-loaded nanocarriers. | ||
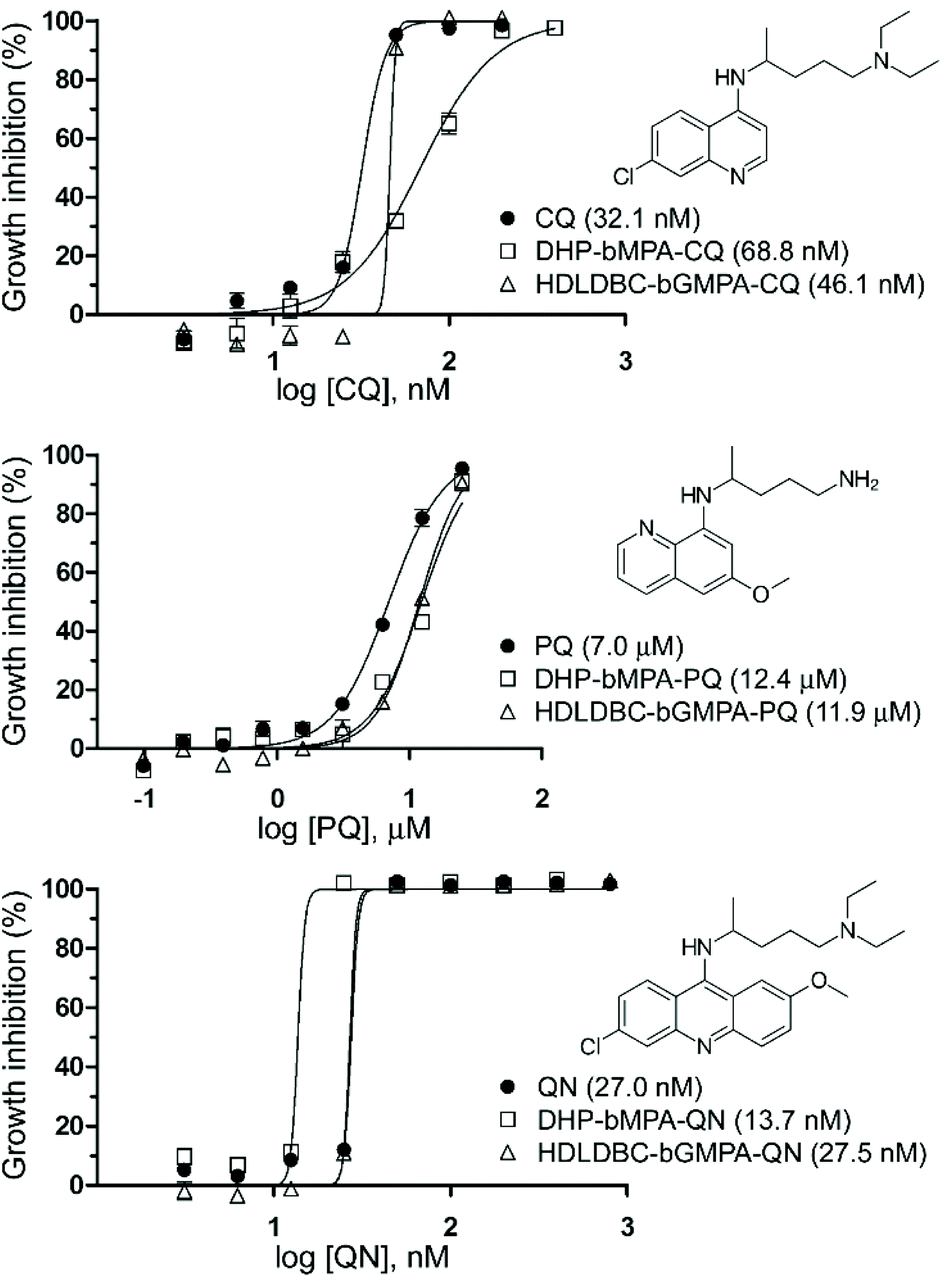 | ||
| Fig. 9 Growth inhibition assays of P. falciparum in vitro cultures treated with CQ, PQ and QN, either free or encapsulated in HDLDBC-bGMPA and DHP-bMPA. In parentheses are indicated the corresponding IC50 values. | ||
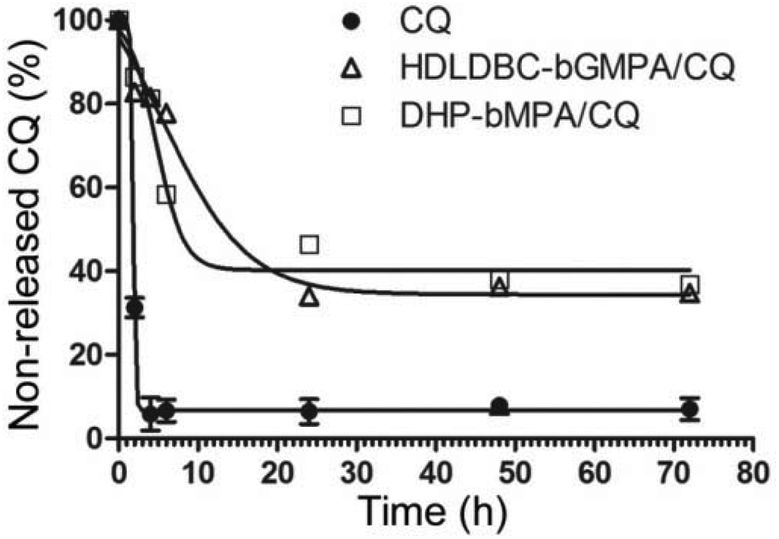 | ||
| Fig. 10 Release profiles of CQ from the dendrimers DHP-bMPA and HDLDBC-bGMPA at 37 °C. | ||
In vivo antimalarial activity of CQ-loaded nanocarriers
MTD assays indicated that DHP-bMPA and HDLDBC-bGMPA could be safely administered to mice without evident signs of animal distress up to respective doses of at least 37.5 and 120 mg kg−1. For the evaluation of the formulations in an in vivo model of malaria, we selected CQ because, being the in vitro performance of its encapsulated form vs. the free compound similar to that of PQ and QN, this drug had been successfully tested in our group as part of different polymeric and liposomal encapsulations.18,35,38 When polymer-CQ conjugates were administered intravenously (1.9 mg CQ per kg per day for 4 consecutive days) to mice infected with the lethal murine malaria parasite P. yoelii yoelii 17XL (PyL) MRA-267, DHP-bMPA-CQ did not improve survival significantly, but animals treated with HDLDBC-bGMPA-CQ survived much longer (3 out of 5 mice lived ≥20 days) than untreated controls, which typically died at day 6 (Fig. 11A and C). One mouse treated with HDLDBC-bGMPA-CQ was actually cured and, when re-infected at day 69 and left untreated, it recovered completely without showing any symptoms of malaria or developing parasitemia above the detection level of microscopy examination. The detection in the plasma of this cured animal of antibodies against P. yoelii antigens (Fig. 11B) was consistent with the development of immunity against the disease. Free CQ-treated controls which were administered the same drug concentration were cured in 4 out of 5 animals, which became also resistant to re-infection. Although HDLDBC-bGMPA-encapsulated CQ did not improve the efficacy of such a good drug as CQ, the strategy presented here can be adapted to the targeted drug delivery of other antimalarials, already existing or yet to be discovered.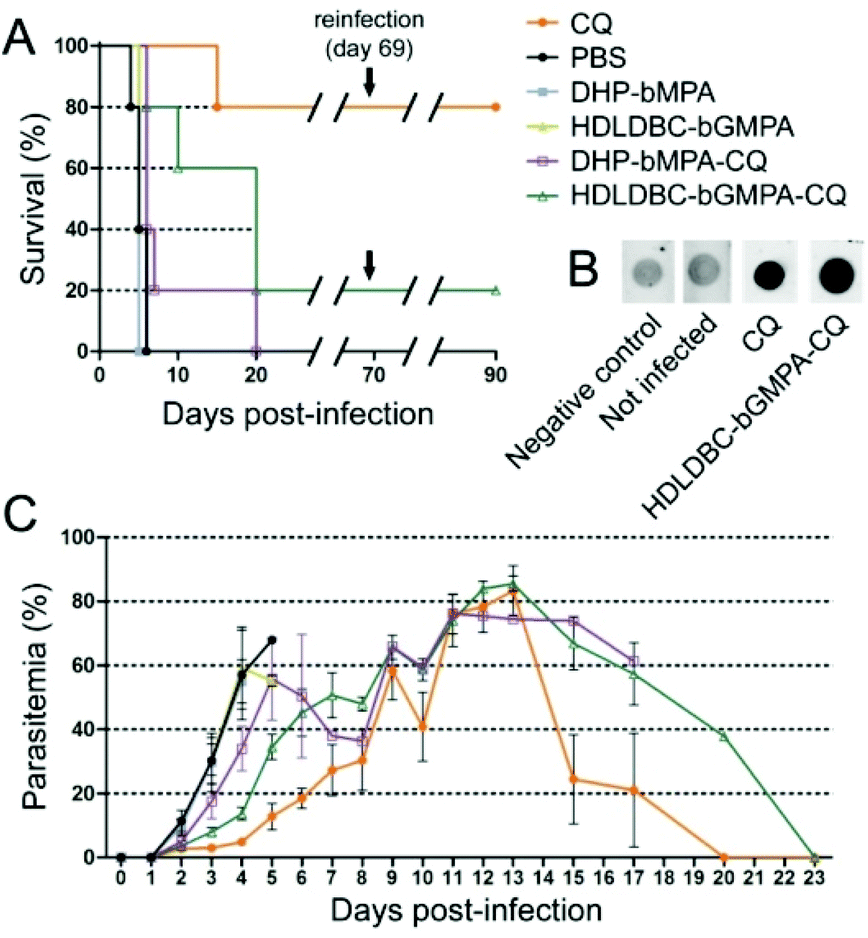 | ||
| Fig. 11 Four-day blood suppressive antimalarial activity assay of free CQ and of carrier-CQ conjugates. (A) Kaplan–Meier plot of the survival of P. yoelii yoelii 17XL (PyL) MRA-267-infected mice treated with 1.9 mg CQ per kg per day for 4 consecutive days, administered intravenously as diphosphate–drug or dendrimer–drug. When all surviving animals had completely cleared Plasmodium infection they were re-infected (on day 69) with P. yoelii and left untreated. (B) Dot blot for the detection at day 90 in the plasma of surviving animals of antibodies against P. yoelii. The negative control was processed substituting PBS for plasma. (C) Parasitemia counts of the different mouse groups until animal death or parasite clearance. | ||
In vivo, HDLDBC-bGMPA-CQ worked better than DHP-bMPA-CQ, possibly because the former targets all RBCs. The loading of antimalarial drugs into non-parasitized red blood cells has been described as an efficient approach to significantly reduce parasite survival,35,36 as long as neither nanocarriers nor drugs affect the natural role of erythrocytes as O2 and CO2 transporters. Both dendrimeric structures studied in this work lack significant in vitro cytotoxicity and hemolytic activity, suggesting that they will not interfere with the red blood cell physiology. HDLDBC-bGMPA then holds promise for the development of innovative antimalarial prophylactic strategies at the cell level whereby Plasmodium would be exposed to drugs since the very first moment after invading a host cell.
The modest in vivo efficacy of the encapsulated formulations relative to the free drug is, in part, likely resulting from renal and splenic clearance of the nanoparticles, in addition to their endothelial cell uptake. Nanoparticles must be larger than 20 nm in diameter to avoid filtration by the kidney,39 and smaller than 100 nm to avoid a specific sequestration by sinusoids in spleen and fenestra of liver, which are approximately 150–200 nm in diameter.40 Therefore, systemically administered nanoparticles should have diameters from 20 to 100 nm,41 and overall, literature suggests that nanoparticles in the 50–100 nm size range display the lowest blood clearance rates.42 Since the average diameter of the dendrimeric nanoparticles used here is just below 20 nm, increasing their size to ca. 50 nm might provide an improved pharmacokinetics and better in vivo performance.
In addition, upon intravenous administration of nanoparticles, these are coated by a variety of serum proteins which are recognized by the scavenger receptor on macrophage cell surfaces and internalized, leading to a significant loss of nanoparticles from the circulation.43 The serum proteins binding on the nanoparticles are also termed “opsonins”, and the macrophages contributing the major loss of injected dose are also known as the reticuloendothelial system or mononuclear phagocyte system. Reducing protein binding is the key point for developing a long-circulation nanoparticle formulation. To minimize opsonization, the most commonly used strategy is to conjugate onto the surface of the nanoparticles the polyethylene glycol polymer44 or polyoxazolines,45 both hydrophilic polymers that provide good steric hindrance for preventing protein binding.
Finally, in addition to expanding blood residence time and reducing unspecific interactions with non-target cells and tissues, a faster targeting dynamics can be conferred to the nanoparticles by coating them with molecules binding pRBCs with certain degree of specificity, as it has been described for heparin.46,47 Here we have shown that although DHP-bMPA exhibited a remarkable preferential binding to pRBCs vs. RBCs, about 40% of pRBCs interacted with this polymer similarly as a fraction of the non-infected erythrocyte population did (Fig. 6). The functionalization of DHP-bMPA-Rho with up to 15% heparin-FITC (w/w) did not affect the targeting specificity of the polymer towards pRBCs (Fig. S3.1†), and it actually favored specific interactions with pRBCs vs. RBCs (data not shown). The known activity of heparin as targeting element of liposomes47 suggests that its presence on the nanocarrier increased the number of polymers bound to each pRBC. Last but not least, since heparin has antimalarial activity, this result indicates that it can be a constituent of pRBC-targeted multicomponent nanoparticles carrying different types of antimalarial agents.
Conclusions
To eradicate malaria, there is an urgent need for the combination of different strategies working together, because it is unlikely for a single approach to be capable of success. It is important to emphasize the importance of implementing in advance efficient methods of antimalarial drug encapsulation and targeted delivery in order to make a good use of future therapeutic compounds. This should result in a more rational administration regime that could contribute to expand the time during which drugs can be used before resistance evolves. In addition, such strategy might widen the number of chemicals that reach the clinic if eventual unspecific toxicities can be reduced through a targeted delivery approach. The resulting lower overall doses should not trigger pernicious side-effects in the patient while keeping high local doses on the parasite in order to quickly eliminate all the cells of the pathogen, thus reducing the evolution of resistances. Here, we have described micellar carriers based on dendritic macromolecules containing bis-MPA and glycine that hold promise for the development of future antimalarial nanomedicines targeted to both Plasmodium-infected and non-infected erythrocytes.Authors contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by grants BIO2014-52872-R, CTQ2015-70174-P, MAT2015-66208-C3-1-P (Ministerio de Ciencia, Innovación y Universidades, Spain, including FEDER funds), 2014-SGR-938 (Generalitat de Catalunya, Spain), and (Gobierno de Aragón-FSE). ISGlobal and IBEC are members of the CERCA Programme, Generalitat de Catalunya. A. L. thanks the Ministerio de Ciencia, Innovación y Universidades for his grant (FPU12/05210). L. N. B.-C. is supported by the European Commission under Horizon 2020's Marie Skłodowska-Curie Actions COFUND scheme (Grant Agreement no. 712754) and by the MINECO's Severo Ochoa programme (Grant SEV-2014-0425 (2015–2019)). The authors would like to acknowledge the use of the Servicios Científico Técnicos of CEQMA (Universidad de Zaragoza-CSIC) and of the LMA (INA-Universidad de Zaragoza). The authors acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).Notes and references
- World Health Organization, World Malaria Report 2018. http://www.who.int/malaria/publications/world-malaria-report-2018/report/en/, 2018.
- V. S. Moorthy, R. D. Newman, P. Duclos, J. M. Okwo-Bele and P. G. Smith, Lancet Infect. Dis., 2013, 13, 280–282 CrossRef CAS PubMed.
- A. Mbengue, S. Bhattacharjee, T. Pandharkar, H. Liu, G. Estiu, R. V. Stahelin, S. S. Rizk, D. L. Njimoh, Y. Ryan, K. Chotivanich, C. Nguon, M. Ghorbal, J. J. Lopez-Rubio, M. Pfrender, S. Emrich, N. Mohandas, A. M. Dondorp, O. Wiest and K. Haldar, Nature, 2015, 520, 683–687 CrossRef CAS PubMed.
- P. L. Alonso and M. Tanner, Nat. Med., 2013, 19, 150–155 CrossRef CAS PubMed.
- M. Prudêncio, A. Rodriguez and M. M. Mota, Nat. Rev. Microbiol., 2006, 4, 849–856 CrossRef PubMed.
- A. F. Cowman and B. S. Crabb, Cell, 2006, 124, 755–766 CrossRef CAS PubMed.
- K. S. Griffith, L. S. Lewis, S. Mali and M. E. Parise, J. Am. Med. Assoc., 2007, 297, 2264–2277 CrossRef CAS PubMed.
- R. G. Feachem, A. A. Phillips, G. A. Targett and R. W. Snow, Lancet, 2010, 376, 1517–1521 CrossRef.
- P. L. Alonso, Int. Microbiol., 2006, 9, 83–93 Search PubMed.
- J. P. Daily and J. Clin, Pharmacol., 2006, 46, 1487–1497 CAS.
- European Science Fundation: ESF Forward Look on Nanomedicine 2005. http://www.nanopharmaceuticals.org/files/nanomedicine.pdf, 2005.
- M. Saltzman and T. Desai, Ann. Biomed. Eng., 2006, 34, 270–275 CrossRef PubMed.
- P. Urbán, J. J. Valle-Delgado, E. Moles, J. Marqués, C. Díez and X. Fernàndez-Busquets, Curr. Drug Targets, 2012, 13, 1158–1172 CrossRef.
- N. Kuntworbe, N. Martini, J. Shaw and R. Al-Kassas, Drug Dev. Res., 2012, 73, 167–184 CrossRef CAS.
- P. Urbán and X. Fernàndez-Busquets, Curr. Med. Chem., 2014, 21, 605–629 CrossRef.
- M. Selin, L. Peltonen and L. M. J. Bimbo, J. Drug Delivery Sci. Technol., 2016, 34, 10–20 CrossRef CAS; H. Yang, Nanomedicine, 2016, 12, 309 CrossRef PubMed; A. M. Caminade and C. O. Turrin, J. Mater. Chem. B, 2014, 2, 4055–4066 RSC; P. Kesharwani, K. Jain and N. K. Jain, Prog. Polym. Sci., 2014, 39, 268–307 CrossRef.
- S. García-Gallego, A. M. Nyström and M. Malkoch, Prog. Polym. Sci., 2015, 48, 85–110 CrossRef.
- J. Movellan, P. Urbán, E. Moles, J. M. De la Fuente, T. Sierra, J. L. Serrano and X. Fernàndez-Busquets, Biomaterials, 2014, 35, 7940–7950 CrossRef CAS PubMed.
- R. D. Sikwal, R. S. Kalhapure and T. Govender, Eur. J. Pharm. Sci., 2017, 97, 113–134 CrossRef PubMed.
- B. S. Bolu, R. Sanyal and A. Sanyal, Molecules, 2018, 23, 1570–1595 CrossRef PubMed.
- I. D. Goodyer, B. Pouvelle, T. G. Schneider, D. P. Trelka and T. F. Taraschi, Mol. Biochem. Parasitol., 1997, 87, 13 CrossRef CAS PubMed.
- A. Fonseca, M. A. Gil and P. M. Simões, Prog. Polym. Sci., 2014, 39, 1291–1311 CrossRef CAS.
- M. Winnacker and B. Rieger, Polym. Chem., 2016, 7, 7039–7046 RSC.
- A. Lancelot, R. González-Pastor, R. Clavería-Gimeno, P. Romero, O. Abián, P. Martín-Duque, J. L. Serrano and T. Sierra, J. Mater. Chem. B, 2018, 6, 3956–3968 RSC.
- A. Lancelot, R. González-Pastor, A. Concellón, T. Sierra, P. Martín-Duque and J. L. Serrano, Bioconjugate Chem., 2017, 28, 1135–1150 CrossRef CAS PubMed.
- M. C. A. Stuart, J. C. van de Pas and J. B. F. N. Engberts, J. Phys. Org. Chem., 2005, 18, 929–934 CrossRef CAS.
- I. Jiménez-Pardo, R. González-Pastor, A. Lancelot, R. Clavería-Gimeno, A. Velázquez-Campoy, O. Abián, M. B. Ros and T. Sierra, Macromol. Biosci., 2015, 15, 1381–1391 CrossRef PubMed.
- P. Urbán, J. Estelrich, A. Cortés and X. Fernàndez-Busquets, J. Controlled Release, 2011, 151, 202–211 CrossRef PubMed.
- S. L. Cranmer, C. Magowan, J. Liang, R. L. Coppel and B. M. Cooke, Trans. R. Soc. Trop. Med. Hyg., 1997, 91, 363–365 CrossRef CAS PubMed.
- C. Lambros and J. P. Vanderberg, J. Parasitol., 1979, 65, 418–420 CrossRef CAS PubMed.
- D. A. Fidock, P. J. Rosenthal, S. L. Croft, R. Brun and S. Nwaka, Nat. Rev. Drug Discovery, 2004, 3, 509–520 CrossRef CAS PubMed.
- J. Marques, E. Vilanova, P. A. S. Mourão and X. Fernàndez-Busquets, Sci. Rep., 2016, 6, 24368 CrossRef CAS PubMed.
- G. R. Newkome, C. N. Moorefield, G. R. Baker, M. J. Saunders and S. H. Grossman, Angew. Chem., Int. Ed. Engl., 1991, 30, 1178–1180 CrossRef.
- X. Fan, Z. Lia and X. J. Loh, Polym. Chem., 2016, 7, 5898–5919 RSC.
- E. Moles, P. Urbán, M. B. Jiménez-Díaz, S. Viera-Morilla, I. Angulo-Barturen, M. A. Busquets and X. Fernàndez-Busquets, J. Controlled Release, 2015, 210, 217–229 CrossRef CAS PubMed.
- E. Moles and X. Fernàndez-Busquets, Future Med. Chem., 2015, 7, 837–840 Search PubMed.
- E. Moles, S. Galiano, A. Gomes, M. Quiliano, C. Teixeira, I. Aldana, P. Gomes and X. Fernàndez-Busquets, Biomaterials, 2017, 145, 178–191 CrossRef CAS PubMed.
- P. Urbán, J. J. Valle-Delgado, N. Mauro, J. Marques, A. Manfredi, M. Rottmann, E. Ranucci, P. Ferruti and X. Fernàndez-Busquets, J. Controlled Release, 2014, 177, 84–95 CrossRef PubMed.
- D. Venturoli and B. Rippe, Am. J. Physiol.: Renal Physiol., 2005, 288, F605–F613 CrossRef CAS PubMed.
- X. Duan and Y. Li, Small, 2012, 9, 1521–1532 CrossRef PubMed.
- M. E. Davis, Z. Chen and D. M. Shin, Nat. Rev. Drug Discovery, 2008, 7, 771–782 CrossRef CAS PubMed.
- F. Alexis, E. Pridgen, L. K. Molnar and O. C. Farokhzad, Mol. Pharm., 2008, 5, 505–515 CrossRef CAS PubMed.
- M. C. Woodle, M. S. Newman and J. A. Cohen, J. Drug Targeting, 1994, 2, 397–403 CrossRef CAS PubMed.
- S. D. Li and L. Huang, Mol. Pharm., 2008, 5, 496–504 CrossRef CAS PubMed.
- R. B. Restani, J. Conde, R. F. Pires, P. Martins, A. R. Fernandes, P. V. Baptista, V. D. B. Bonifácio and A. Aguiar-Ricardo, Macromol. Biosci., 2015, 15, 1045–1051 CrossRef CAS PubMed.
- X. Fernàndez-Busquets, Future Med. Chem., 2013, 5, 737–739 CrossRef PubMed.
- J. Marques, E. Moles, P. Urbán, P. Prohens, M. A. Busquets, C. Sevrin, C. Grandfils and X. Fernàndez-Busquets, Nanomedicine, 2014, 10, 1719–1728 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8bm01600c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |