
Redox-responsive interleukin-2 nanogel specifically and safely promotes the proliferation and memory precursor differentiation of tumor-reactive T-cells†
Yu-Qing
Xie
 a,
Hacer
Arik
a,
Lixia
Wei
b,
Yiran
Zheng
c,
Heikyung
Suh
c,
Darrell J.
Irvine
cd and
Li
Tang
*ab
a,
Hacer
Arik
a,
Lixia
Wei
b,
Yiran
Zheng
c,
Heikyung
Suh
c,
Darrell J.
Irvine
cd and
Li
Tang
*ab
aInstitute of Bioengineering, École polytechnique fédérale de Lausanne (EPFL), Lausanne, Switzerland CH-1015. E-mail: li.tang@epfl.ch
bInstitute of Materials Science & Engineering, École polytechnique fédérale de Lausanne (EPFL), Lausanne, Switzerland CH-1015
cDavid H. Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA
dHoward Hughes Medical Institute, Chevy Chase, Maryland 20815, USA
First published on 22nd January 2019
Abstract
Interleukin-2 (IL-2) is a potent T-cell mitogen that can adjuvant anti-cancer adoptive T-cell transfer (ACT) immunotherapy by promoting T-cell engraftment. However, the clinical applications of IL-2 in combination with ACT are greatly hindered by the severe adverse effects such as vascular leak syndrome (VLS). Here, we developed a synthetic delivery strategy for IL-2 via backpacking redox-responsive IL-2/Fc nanogels (NGs) to the plasma membrane of adoptively transferred T-cells. The NGs prepared by traceless chemical cross-linking of cytokine proteins selectively released the cargos in response to T-cell receptor activation upon antigen recognition in tumors. We found that IL-2/Fc delivered by T-cell surface-bound NGs expanded transferred tumor-reactive T-cells 80-fold more than the free IL-2/Fc of an equivalent dose administered systemically and showed no effects on tumor-infiltrating regulatory T-cell expansion. Intriguingly, IL-2/Fc NG backpacks that facilitated a sustained and slow release of IL-2/Fc also promoted the CD8+ memory precursor differentiation and induced less T-cell exhaustion in vitro compared to free IL-2/Fc. The controlled responsive delivery of IL-2/Fc enabled the safe administration of repeated doses of the stimulant cytokine with no overt toxicity and improved efficacy against melanoma metastases in a mice model.
Introduction
Adoptive T-cell transfer (ACT), a potent immunotherapy that infuses patient-derived T-cells back to fight cancer, has shown dramatic clinical success particularly in the treatment of several hematological cancers.1 However, the efficacy of ACT against solid tumors has been rather limited to date partially because of the highly immunosuppressive tumor microenvironment (TME).2 Various strategies are actively pursued to support tumor-reactive T-cells and their function3,4 or modulate the TME5,6 with adjuvant agents such as stimulatory cytokines. Among those agents, IL-2 is a promising candidate as it is a potent T-cell growth factor. When given systemically at high doses, IL-2 could stimulate the proliferation and enhance the therapeutic function of adoptively transferred T-cells leading to overall improved efficacy.7 However, the wide clinical applications of IL-2 treatment are largely restricted by severe side effects such as vascular leak syndrome8 and its non-specific stimulation activities.9 In combination therapies of IL-2 and ACT, IL-2 could nonspecifically activate endogenous lymphocytes leading to cytokine release syndrome and autoimmune tissue damages.10 Due to the high expression level of α-chain of IL-2 receptor in regulatory T (Treg)-cells, which has high binding affinity with IL-2, systemically administered IL-2 could significantly expand Treg cells in TME leading to immunosuppression.11 In addition, controlling the dosage of IL-2 also appears critical in T-cell activation as strong IL-2 signal is known to drive CD8+ T-cells to become terminally differentiated effector cells that are destined for rapid cell death.12 Therefore, it is crucial to focus the activity of IL-2 on adoptively transferred T-cells in tumor and precisely control the doses.In order to improve the delivery of adjuvant agents for ACT, we previously developed a T-cell receptor (TCR)-signaling responsive nanoparticle backpack for the targeted delivery of IL-15 as an adjuvant cytokine for enhanced ACT.13 Here, we extend this approach for the delivery of IL-2 to address the aforementioned challenges in using IL-2 for supporting ACT. A redox responsive nanogel (NG) prepared through chemical cross-linking of IL-2/Fc, a fusion protein between murine IL-2 and a mutated antibody Fc domain,14 was backpacked onto the plasma membrane of donor T-cells, enabling controlled pseudoautocrine release of IL-2/Fc leading to the specific stimulation of transferred T-cells. We found that IL-2/Fc delivered by T-cell surface-bound NGs expanded transferred tumor-reactive T-cells 80-fold more than the systemically administered free IL-2/Fc of an equivalent dose and showed no effects on tumor-infiltrating Treg cell expansion. Interestingly, T-cell surface-bound IL-2/Fc NGs also promoted CD8+ memory precursor differentiation and induced less T-cell exhaustion in vitro compared to free IL-2/Fc likely due to the sustained and slow release of IL-2/Fc. By achieving specific delivery of IL-2/Fc to adoptively transferred T-cells in tumor, IL-2/Fc NG backpacks substantially reduced the toxicities, which were observed when the free IL-2/Fc was administered systemically in tandem with ACT in mice and improved the efficacy against melanoma metastases.
Materials and methods
Materials
IL-2/Fc, a bivalent fusion protein of mouse wild-type IL-2 with the C terminus linked to a mouse IgG2a backbone, was a generous gift from Dane Wittrup's lab (MIT, MA, USA) and was prepared as described previously.14 Amine-Poly(ethylene glycol)-amine (NH2-PEG10k-NH2) was purchased from Laysan Bio (Arab, AL, USA). Polyethylene glycol-b-polylysine (PEG5k-PLL33k) was purchased from Alamanda Polymers (Huntsville, AL, USA). Bis(sulfosuccinimidyl) suberate (BS3 linker) was purchased from Thermo Fisher Scientific (Waltham, MA, USA). Retronectin Recombinant Human Fibronectin Fragment was from Clontech (Mountain View, CA). All other chemicals and solvents were purchased from Sigma-Aldrich (St Louis, MO, USA) unless otherwise noted. All reagents were used as received unless otherwise noted.Mice and cell lines
Experimental procedures in mouse studies were approved by the Swiss authorities (Canton of Vaud, animal protocol ID 3206) and performed in accordance with EPFL CPG guidelines. Six- to eight-week-old female Thy1.2+ C57Bl/6 mice, TCR-transgenic Thy1.1+ pmel-1 mice (B6.Cg-Thy1a/Cy Tg(TcraTcrb)8Rest/J) were purchased from The Jackson Laboratory (Bar Harbor, ME, USA) or Charles River Laboratories (Lyon, France) and maintained in the animal facility. B16F10 melanoma cells were acquired from the American Type Culture Collection (Manassas, VA, USA) and cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% Fetal Bovine Serum (FBS) (Gibco, Waltham, MA, USA).Synthesis of IL-2/Fc NGs
Synthesis of IL-2/Fc NG was similar as that of IL-15Sa NG as reported previously.13 First, the redox responsive cross-linker NHS-SS-NHS (Fig. 1) was synthesized as previously reported and dissolved in anhydride DMSO at a concentration of 10 μg μL−1.13 Next, NHS-SS-NHS (73.4 μg, 0.168 μmol, 15 equiv.) dissolved in 7.34 μL DMSO was added to a IL-2/Fc (1000 μg, 0.0112 μmol, 1 equiv.) solution in 100 μL PBS pH 7.4. The mixture was rotated at 25 °C for 30 min followed by the addition of 893 μL PBS buffer. NH2-PEG10k-NH2 (561 μg, 0.0561 μmol, 5 equiv.) in 28.1 μL PBS buffer was then added to the diluted solution. The reaction mixture was rotated at 25 °C for another 30 min. The resultant NGs were then washed with PBS (500 μL × 3) in an Amicon® Ultra Centrifugal Filters (molecular weight cut-off 100 kDa) (Millipore, Billerica, MA, USA). Non-degradable IL-2/Fc NGs were prepared similarly except using a permanent cross-linker, bis(sulfosuccinimidyl) suberate (BS3, 30 equiv.) (Fig. S1†). To prepare fluorescently labeled NGs, IL-2/Fc were fluorescently labeled with Alexa Fluor 647 NHS ester (Thermo Fisher Scientific) and purified with Amicon® Ultra Centrifugal Filters (molecular weight cut-off 50 kDa). Fluorescent IL-2/Fc (10 mol%) was mixed with non-labeled IL-2/Fc for the preparation of fluorescent NGs following the same procedure as described above. | ||
| Fig. 1 Schematic illustration of the synthesis of IL-2/Fc nanogels (NGs) and backpacking NGs on T-cell plasma membrane. | ||
Characterizations of IL-2/Fc NGs
IL-2/Fc NG formation and complete reaction was verified by HPLC with a size-exclusion column (BioBasic™ SEC 300 LC Column, Thermo Fisher Scientific). The NG formulation was also confirmed in sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) by running samples in MOPS buffer (Invitrogen, Carlsbad, CA, USA). The gel was stained with Coomassie Brillant Blue G250 (Fluka, St Gallen, Switzerland). The reduction-triggered release was induced by a reducing agent (Invitrogen) containing dithiothreitol (DTT, 50 mM). The morphology and dry sizes of NGs were determined by transmission electron microscopy (FEI Tecnai, Hillsboro, OR, USA). A NG solution in deionized water (0.5 mg mL−1) was used for the measurement of the hydrodynamic sizes with dynamic light scattering (Malvern Zetasizer, Malvern Instruments Ltd, Worcestershire, UK). The final concentrations of NGs were determined with a NanoDrop 1000 Spectrophotometer (Thermo Fisher Scientific).Release kinetics of IL-2/Fc from NGs
The IL-2/Fc NGs were dispersed in Hank's balanced salt solution (HBSS, Gibco) (0.1 mg mL−1) and incubated at 37 °C in the presence of glutathione (GSH) with different concentrations. At selected time points, replicates of solution were analyzed with HPLC equipped with a size-exclusion column to determine the percentage of released IL-2/Fc. The biological activity of released IL-2/Fc was verified by T-cell expansion studies described below.Activation of pmel-1 CD8+ T-cells
Spleens from pmel-1 Thy1.1+ mice were ground through a 70 μm cell strainer (Fisher Scientific, Pittsburgh, PA, USA), and red blood cells were lysed by incubation with ACK lysis buffer (Gibco, 2 mL per spleen) for 5 min at room temperature. After washing with PBS and filtered again through a 70 μm cell strainer, splenocytes were cultured in 10% FBS supplemented RPMI 1640 culture medium with the presence of 1 μM human gp10025–33 (GenScript, Nanjing, China), 10 ng mL−1 recombinant mouse IL-2 (PeproTech, Rocky Hill, NJ, United States) and 1 ng mL−1 recombinant mouse IL-7 (PeproTech) for 3 days followed by Ficoll-Pague Plus (GE Health Care, Chicago, IL, USA) gradient separation to eliminate dead cells. After culture in the medium containing recombinant mouse IL-7 (1 ng mL−1) for one more day, the pmel-1 CD8+ T-cells (>95% pure; Fig. S2†) were used for in vitro and in vivo studies.For bioluminescence imaging experiments, click beetle red luciferase (CBR-Luc)5 was introduced into pmel-1 T-cells by retroviral transduction. Phoenix eco viral packaging cells were seeded at 4.0 × 106 cells per 10 cm tissue culture dish in 10 mL DMEM medium containing 10% FBS. After incubation overnight at 37 °C, phoenix cells were exchanged with 10 mL fresh DMEM with 10% FBS, transfected with CBR-Luc plasmid and phoenix eco plasmid using a calcium phosphate transfection kit and cultured at 32 °C for 24 h. DMEM was then replaced with 6 mL RPMI containing 10% FBS and transfected phoenix eco cells were incubated for another 24 h. Supernatant containing the retrovirus-packaged CBR-Luc gene was collected and replaced with fresh RPMI for another 24 h incubation. Supernatant was collected again and combined with that collected 24 h earlier, and sterile filtered (0.45 μm). Six-well non-tissue culture plates (BD Falcon, NY, USA) were coated with 1 mL retronectin (15 μg mL−1) for 18 h at 4 °C, then excess retronectin was aspirated. Activated pmel-1 CD8+ T-cells were suspended in filtered viral sups (RPMI collected previously) with 10 ng mL−1 IL-2 at 1.8 × 106 mL−1, 3 mL was added to each retronection-coated well, and spinoculation was conducted by centrifuging at 2000g for 1 h at 25 °C. Transduced T-cells were then incubated at 37 °C. Six hours later, 1 mL of fresh RPMI was added with 10 ng mL−1 IL-2. Transduced, activated pmel-1 T-cells were used 1 day later for adoptive transfer studies.
Conjugation of IL-2/Fc NGs to T-cell plasma membrane
IL-2/Fc NGs (950 μg, 0.011 μmol) labeled with Alexa Fluor 647 in PBS (950 μL) were first activated with BS3 linker (314 μg, 0.55 μmol) or NHS-SS-NHS (240 μg, 0.55 μmol) for 30 min at room temperature and then collected with Amicon® Ultra Centrifugal Filters (molecular weight cut-off 50 kDa) and washed with PBS (1.5 mL × 3). The activated NGs were diluted with PBS to the concentration of 1 μg μL−1 followed by the addition of PEG5k-b-PLL33k (33.4 μg, 8.8 × 10−4 μmol, 0.08 equiv. of IL-2/Fc) in 33.4 μL PBS. The mixture was rotated at 25 °C for 30 min and used without further purification. The modified IL-2/Fc NGs (950 μg) in 950 μL PBS was added to pmel-1 CD8+ T-cells (95 × 106) in 500 μL HBSS followed by incubation at 37 °C for 1 h. The T-cells with surface-coupled NGs were collected by centrifugation at 800g for 5 min, washed with PBS (1.0 mL × 2) and resuspended in buffer or medium at the desired concentrations for in vitro or in vivo studies. For measurements of NG coupling efficiency, after incubation, the supernatants were collected and measured for fluorescence intensity at excitation and emission wavelengths of 640 nm and 680 nm, respectively by using Varioskan™ LUX multimode microplate reader (Thermo Fisher Scientific). Fluorescence readings were converted to NG concentrations using standard curves prepared from serial dilutions of NG stock solutions. The amount of coupled NG was calculated by subtracting the unbound NG from the total added amount. NG loading per cell was controlled by varying the mass of NGs added to cells for coupling.Release kinetics of proteins from NGs coupled on T-cell surface
Activated pmel-1 CD8+ T-cells were labeled with carboxyfluorescein succinimidyl ester (CFSE). IL-2/Fc NG labeled with Alexa Fluor 647 fluorescence dye was prepared and conjugated to activated pmel-1 CD8+ T-cells as described above. T-cells were incubated in culture medium at 37 °C with or without anti-CD3/CD28-coated beads. Cells were collected at selected time points and analyzed by flow cytometry (Attune NxT, Life Technologies, CA, USA) for the measurement of mean fluorescence intensity (MFI) over time. To calculate the relative NG density on T-cell surface, the MFI of Alexa Fluor 647 fluorescence dye was normalized by the MFI of CFSE.In vitro expansion assay of T-cells
Activated pmel-1 CD8+ T-cells were labeled with CFSE and then conjugated with IL-2/Fc NGs as described above. Control groups include T-cells only, and T-cells with free IL-2/Fc at an equivalent dose. T-cells resuspended in culture medium at a density of 5.0 × 104 cells per mL were collected at indicated time and analyzed with flow cytometry for the cell counts and CFSE dilution.In vivo T-cell expansion
In a pulmonary melanoma metastasis model, B16F10 melanoma cells (1.0 × 106) were intravenously (i.v.) injected into tail veins of C57Bl/6 mice at day 8. Mice were sublethally lymphodepleted by total body irradiation (5 Gy) at day 1. Activated pmel-1 CD8+ T-cells transduced with CBR-Luc (1.0 × 107) alone or with surface-coupled IL-2/Fc NGs in 200 μL PBS were administered by i.v. injection at day 0. In another group, free IL-2/Fc was administered by i.v. injection immediately after ACT at equivalent total doses. The bioluminescence was measured by IVIS system every other day until day 12.In a subcutaneous melanoma model, B16F10 melanoma cells (1.0 × 106) were administered subcutaneously (s.c.) in the right flanks of C57Bl/6 mice at day 0. Primed pmel-1 CD8+ T-cells (1.0 × 107) alone or with surface-coupled NGs in 200 μL PBS were administered by i.v. injection at day 6. In another group, free IL-2/Fc was administered by i.v. injection immediately after ACT at equivalent total doses. To monitor in vivo T-cell expansion, mice were euthanized on day 19 for flow cytometry analyses. Tumors were collected and ground through a 70 μm cell strainer. All cells were washed with flow cytometry buffer (PBS with 2% FBS) followed by aqua live/dead staining. Cells were stained for surface markers (CD8, Thy1.1, CD4, NK1.1) with antibodies followed by fixation and permeabilization with Foxp3/Transcription Factor Staining Buffer Set (eBioscience, San Diego, CA, USA). Cells were then stained intracellularly for Foxp3 (antibody clone MF-14, BioLegend, San Diego, CA, USA). After washing with FACS buffer, cells were resuspended in FACS buffer and analyzed by flow cytometry. Data analysis was performed using FlowJo software (Tree Star, Oregon, USA).
In vivo bioluminescence imaging
D-Luciferin (PerkinElmer, Waltham, MA, USA) suspended in PBS (15 mg mL−1) was injected intraperitoneally (150 mg kg−1) 5 min before acquisitions. Bioluminescence images were collected on a Xenogen IVIS Spectrum Imaging System (Xenogen, Alameda, CA, USA). Living Image Software Version 3.0 (Xenogen) was used to acquire and quantify the bioluminescence imaging data sets.In vivo toxicity study
B16F10 melanoma cells (1.0 × 106) were administered by s.c. injection in the right flanks of C57Bl/6 mice on day 0. At day 6, mice received i.v. injection of PBS, activated pmel-1 CD8+ T-cells (1.0 × 107) alone, or T-cells with free IL-2/Fc (daily i.v. injections at day 6, 7, and 8), or IL-2/Fc NGs at equivalent dose (surface-bound NG backpacks at day 6; non-coupled NGs at day 7 and day 8 through i.v. injections). Body weights were measured every day. Peripheral blood was collected; spleen, liver and lung were excised and weighted at day 9 when the mice were sacrificed. Serum alanine transaminase (ALT) and aspartate transaminase (AST) activity was measured using Stanbio reagents (Stanbio, Boerne, TX, USA). Organs were fixed in 4% paraformaldehyde (PFA) overnight followed by imbedding in paraffin blocks. Paraffin slides were sectioned at the thickness of 4 μm, and stained with Hematoxylin and Eosin (H&E) for pathological analysis.In vivo therapy study
B16F10 melanoma cells (1.0 × 106) were i.v. injected into tail veins of C57Bl/6 mice at day 0. Mice were sublethally lymphodepleted by total body irradiation (5 Gy) at day 7. Activated pmel-1 CD8+ T-cells (1.0 × 107) alone or with surface-coupled IL-2/Fc NGs in 200 μL PBS were administered by i.v. injection at day 8. In another group, free IL-2/Fc was administered by i.v. injection immediately after ACT at equivalent total dose. Mice were sacrificed at day 20 to evaluate melanoma lung metastasis. Lungs were perfused with PBS and fixed in 4% PFA overnight followed by paraffin blocking, sectioning, and H&E staining.Statistical analysis
Statistical analysis was performed using GraphPad Prism 7 (GraphPad software, Inc., La Jolla, CA, USA). Data are presented as mean ± s.e.m. unless otherwise indicated. Comparisons of two groups were performed by using two-tailed unpaired Student's t test. Comparisons of multiple groups at a single time point were performed by using one-way analysis of variance (ANOVA), and comparisons of multiple groups over time were performed by using two-way ANOVA tests. If statistically significant, p-values were presented as: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.Results and discussion
Preparation and characterizations of IL-2/Fc NGs
We used IL-2/Fc that possesses the prolonged in vivo half-life compared to native IL-2 to prepare a redox-responsive NG (Fig. 1). A disulphide bond-containing bis-N-hydroxy succinimide (NHS) cross-linker (NHS-SS-NHS) was synthesized as we previously reported and utilized to cross-link the cargo proteins through NHS-amine conjugation reaction.13 Consistent with expectations, IL-2/Fc NGs formed at the conditions with optimized cross-linker-to-protein mole ratio (15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and protein concentration (10 μg μL−1) in pH = 7.4 buffer. The amine conjugation reaction was highly efficient with almost all the IL-2/Fc protein molecules cross-linked quantitatively (Fig. 2a and b) and a high loading capacity.13 Minimum amount of non-cross-linked IL-2/Fc or its oligomers can be removed in filtration. The resultant IL-2/Fc NGs were relatively homogeneous in size exhibiting a dry diameter around 50 nm and a hydrodynamic diameter of 100.2 ± 3.5 nm with a poly(ethylene glycol) (PEG) layer (Fig. 1 and 2c, d). To validate the cross-linking through a redox-responsive linker, IL-2/Fc NGs were treated with a reducing agent dithiothreitol (DTT). The as-prepared NGs were completely degraded into soluble protein molecules, which are essentially the same as degraded species from soluble IL-2/Fc treated with the same agent (Fig. 2b). The cross-linker was designed to facilitate the traceless release of native IL-2/Fc through a self-immolative reaction upon cleavage of the disulphide bond exhibiting a fully reversible chemical modification to ensure the intact structure and activity of the released protein (Fig. S3†). To mimic the reductive environment on T-cell surface, IL-2/Fc NGs were incubated with glutathione (GSH) at different concentrations. The cumulative release was accelerated with increased concentrations of GSH in a dose-dependent manner confirming the redox responsiveness of NGs (Fig. 2e).
:1) and protein concentration (10 μg μL−1) in pH = 7.4 buffer. The amine conjugation reaction was highly efficient with almost all the IL-2/Fc protein molecules cross-linked quantitatively (Fig. 2a and b) and a high loading capacity.13 Minimum amount of non-cross-linked IL-2/Fc or its oligomers can be removed in filtration. The resultant IL-2/Fc NGs were relatively homogeneous in size exhibiting a dry diameter around 50 nm and a hydrodynamic diameter of 100.2 ± 3.5 nm with a poly(ethylene glycol) (PEG) layer (Fig. 1 and 2c, d). To validate the cross-linking through a redox-responsive linker, IL-2/Fc NGs were treated with a reducing agent dithiothreitol (DTT). The as-prepared NGs were completely degraded into soluble protein molecules, which are essentially the same as degraded species from soluble IL-2/Fc treated with the same agent (Fig. 2b). The cross-linker was designed to facilitate the traceless release of native IL-2/Fc through a self-immolative reaction upon cleavage of the disulphide bond exhibiting a fully reversible chemical modification to ensure the intact structure and activity of the released protein (Fig. S3†). To mimic the reductive environment on T-cell surface, IL-2/Fc NGs were incubated with glutathione (GSH) at different concentrations. The cumulative release was accelerated with increased concentrations of GSH in a dose-dependent manner confirming the redox responsiveness of NGs (Fig. 2e).
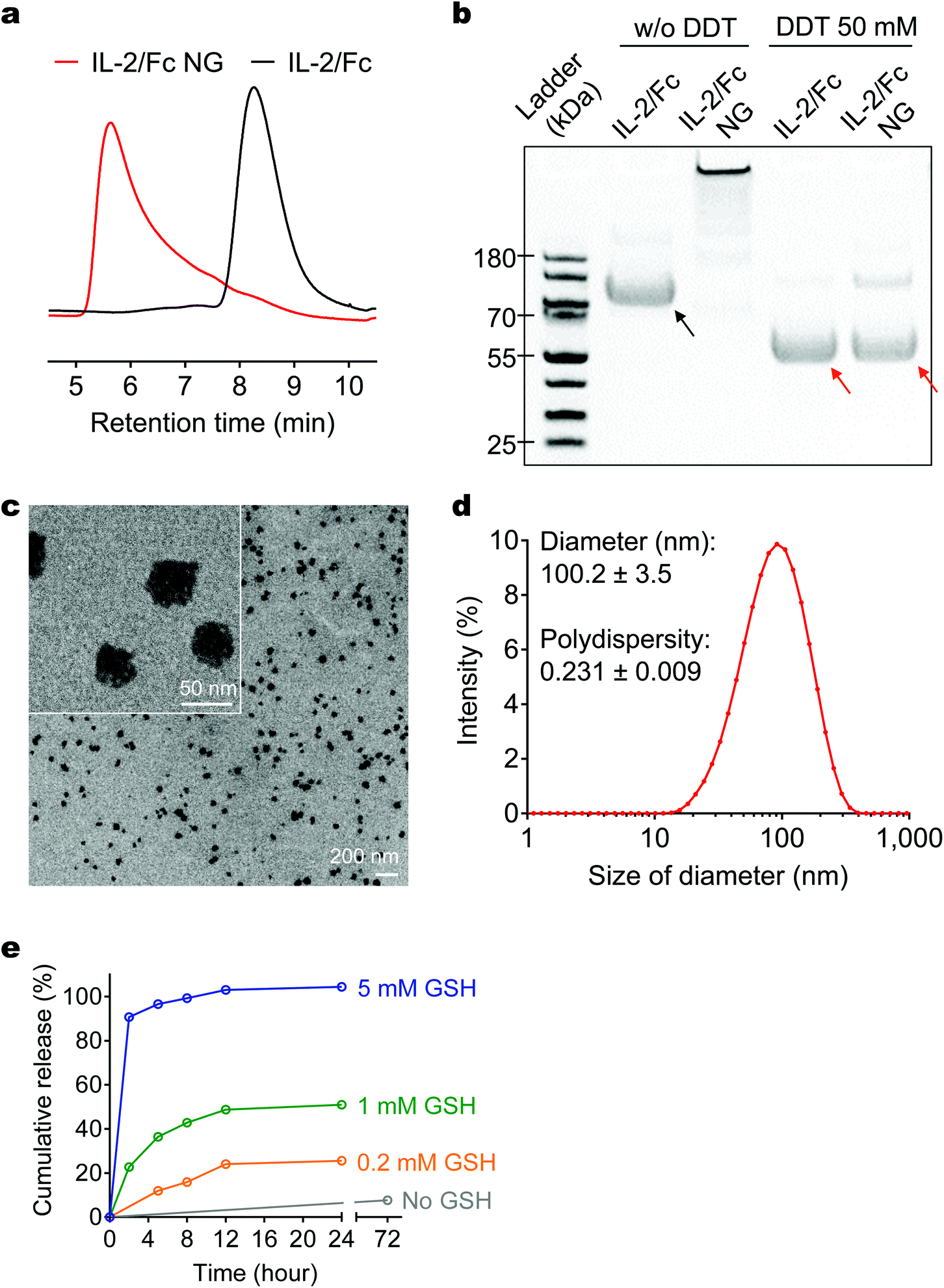 | ||
| Fig. 2 Characterizations of IL-2/Fc NGs. (a) Representative high-performance liquid chromatography (HPLC) analysis of native IL-2/Fc and IL-2/Fc NGs with a size-exclusion column. (b) Representative sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analysis of native IL-2/Fc and IL-2/Fc NGs with or without (w/o) a reducing agent, dithiothreitol (DDT). Black arrow: IL-2/Fc homodimer; Red arrows: IL-2/Fc monomer (disulfide linkage in the hinge region was cleaved by DTT to release monomers). (c) Representative transmission electron microscopy (TEM) images of and IL-2/Fc NGs. (d) Hydrodynamic diameter and polydispersity of IL-2/Fc NGs were determined by dynamic light scattering measurement. (e) Accumulative release of IL-2/Fc from IL-2/Fc NGs in buffers with glutathione (GSH) at different concentrations. | ||
Conjugation of IL-2/Fc NGs on T-cell surface
Next, we covalently conjugated the IL-2/Fc NGs to the amine groups on plasma membrane of T-cells using an amine-reactive linker (bis(sulfosuccinimidyl) suberate or NHS-SS-NHS) (Fig. 1). In order to increase the efficiency of coupling, poly(ethylene glycol)-b-poly(L-lysine) (PEG-PLL), a positively charged copolymer with a PEG segment minimizing non-specific protein binding, was incorporated onto the surface of NG. Covalent conjugation together with the electrostatic interaction facilitated the highly efficient surface backpacking of NGs on T-cells (Fig. 3a; Fig. S4,† average efficiency 74.5 ± 5.1%). The density of NGs bound on T-cell surface could be modularly controlled by using different feeding amount of NGs permitting tunable dosages of adjuvant drugs to be backpacked on T-cells (Fig. 3b). We next assessed the release kinetics of IL-2/Fc from NGs that were fluorescently labeled and backpacked on T-cell surface by monitoring the decay of fluorescence intensity of T-cells with flow cytometry (Fig. 3c). IL-2/Fc NGs bound on T-cell surface exhibited greatly accelerated release of cytokine molecules upon stimulation with anti-CD3/CD28 beads, whereas there was almost no release where a stimulation is lacking. As a comparison, the non-degradable IL-2/Fc NGs showed minimum release even with a stimulation indicating the release of cytokines was triggered by the increased local redox microenvironment (Fig. 3c). Upon the triggering of TCR signaling T-cells largely upregulated the surface reduction activity leading to the redox-responsive release of IL-2/Fc from surface-bound NGs.13 Therefore, the release of adjuvant cytokines from NG backpacks was controlled by the TCR triggering.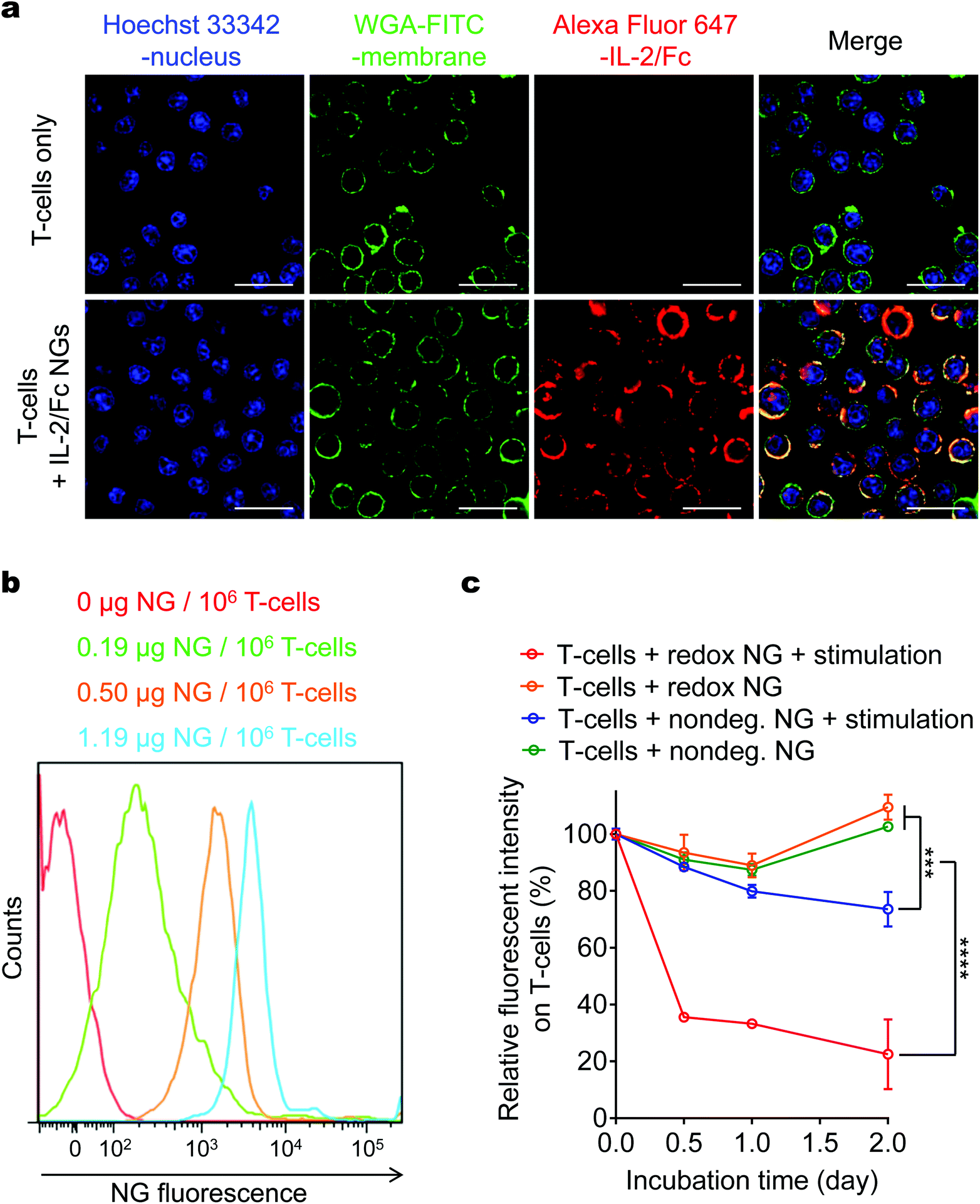 | ||
| Fig. 3 Backpacking IL-2/Fc NGs on T-cell surface. (a) Representative confocal microscopy images of activated pmel-1 CD8+ T-cells with or without fluorescently labeled IL-2/Fc NG backpacks (Alex Fluor 647, red). Nucleus was stained with Hoechst 33342 (blue); cell membrane was stained with WGA-FITC (green). Scale bars: 20 μm. (b) Representative flow cytometric analysis of fluorescent intensity of NGs backpacked on activated pmel-1 CD8+ T-cells at indicated cytokine doses. (c) Flow cytometric analysis of the decay of fluorescence on activated pmel-1 CD8+ T-cells backpacked with fluorescently labeled IL-2/Fc NGs in culture. Data represent the mean ± s.e.m. (n = 3 per group). *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 by Two-Way ANOVA and Tukey's tests. | ||
IL-2/Fc NG backpacks expand T-cells in vitro with increased CD8+ memory precursor differentiation and reduced exhaustion
Surface-bound IL-2/Fc NGs expanded T-cells in vitro to the similar extent as free soluble IL-2/Fc (Fig. 4a and b). Slightly less T-cell expansion in the culture with IL-2/Fc NGs was likely due to the slow release of IL-2/Fc from NGs. We next examined the memory and exhaustion phenotypes of the T-cells after 4-day culture in vitro. Interestingly, we found the expression of CD127, a receptor for IL-7 signaling as well as an essential marker for memory precursors,15 was markedly upregulated in T-cells with surface-bound IL-2/Fc NGs compared to that with soluble IL-2/Fc (Fig. 4c). Additionally, KLRG-1, a marker of terminal effector CD8+ T-cells, was substantially downregulated in NG-backpacked T-cells (Fig. 4d). When IL-7 was supplemented in the culture to mimic the physiological conditions, T-cells with surface-bound IL-2/Fc NGs showed much higher sensitivity to IL-7 signaling and expanded more rapidly compared to T-cells cultured in the presence of soluble IL-2/Fc (Fig. S5†). Together, the results suggest that delivery of IL-2/Fc with the NG backpacks could promote the differentiation of T-cells towards memory precursors. It has been reported that low concentration of IL-2 favored the generation of memory precursors.16 The slow and controlled release of IL-2/Fc from NG backpacks resulted in a low effective concentration of IL-2/Fc around T-cells and thus drove the memory formation. Moreover, T-cells backpacked with IL-2/Fc NGs showed 7.8-fold lower frequency of PD-1+LAG-3+ exhaustion phenotype (Fig. 4e and f). Collectively, IL-2/Fc NG backpacks effectively expanded T-cells in vitro with augmented differentiation toward memory precursor and reduced exhaustion, which are favorable properties of T-cells for potentially enhanced antitumor efficacy.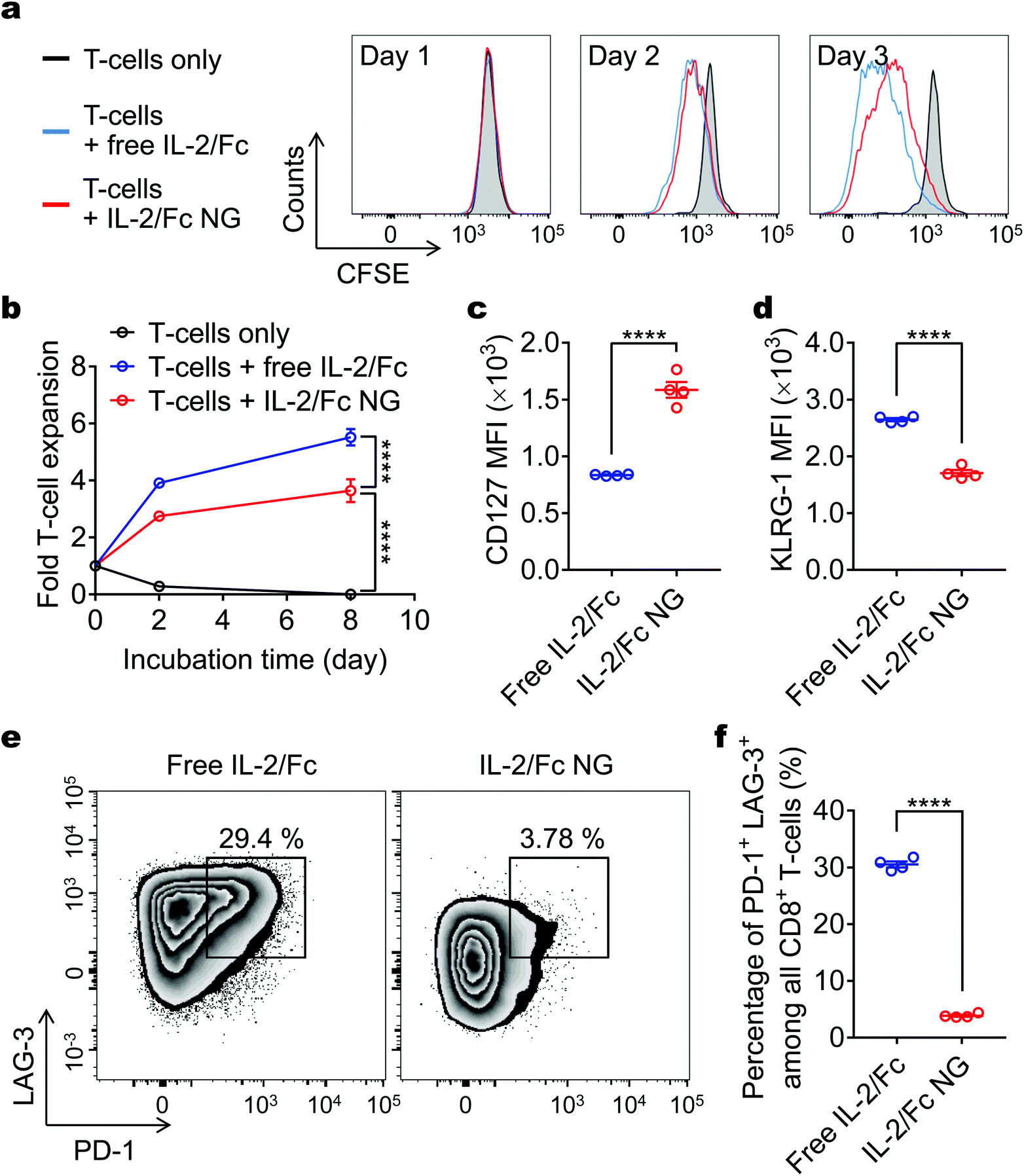 | ||
| Fig. 4 IL-2/Fc NG backpacks promote CD8+ T-cell expansion and memory precursor formation in vitro. Carboxyfluorescein succinimidyl ester (CFSE)-labeled activated pmel-1 CD8+ T-cells (1.0 × 104) were cultured alone or in the presence of free IL-2/Fc (0.1 μg) or IL-2/Fc NG backpacks (0.1 μg) for 8 days. (a, b) Representative flow cytometric analysis of T-cell divisions at day 3 (a) and fold change of cell number over time (b) is shown. (c, d) Mean fluorescence intensity (MFI) of CD127 (c) and KLRG-1 (d) expressed on T-cells at day 4. (e, f) Flow cytometric analysis of the expression level of PD-1 and LAG-3 of CD8+ T-cells (e) and the percentage of PD-1high LAG-3high exhausted T-cells (f) at day 4. Data represent the mean ± s.e.m. (n = 4 per group). *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 by Two-Way ANOVA and Tukey's tests or two-tailed unpaired Student's t test. | ||
IL-2/Fc NG backpacks potently and selectively expand the tumor-reactive T-cells
We next assessed the efficiency and specificity of T-cell expansion by IL-2/Fc NG backpacks in vivo. In a metastatic tumor model, mouse melanoma B16F10 cells were inoculated i.v. to induce tumor nodules in the lungs. Pmel-1 TCR transgenic gp100-specific CD8+ T-cells17 transduced with luciferase were adoptively transferred to the mice preconditioned with lymphodepletion one day before ACT. The expansion of transferred-cells was monitored in vivo using bioluminescence imaging (Fig. 5a and b). At an equivalent dose, IL-2/Fc NG backpack increased the overall T-cell expansion in the whole body of mice 2.5-fold more than that with i.v. administered soluble IL-2/Fc at day 8 post adoptive transfer (Fig. 5c). By focusing the activity of IL-2/Fc around the transferred T-cells, NG backpacks markedly increased the effectiveness of the delivered cytokine adjuvant. However, after reaching the peak value, the overall expansion of T-cells started to decrease on day 10 likely due to a contraction phase of T-cell proliferation following the reaction with target cells and the immunosuppression in TME. In contrast, a single dose of free IL-2/Fc immediately after ACT showed minimal effects on enhancing the expansion of tumor-reactive T-cells over the course of the study presumably due to the fast systemic dissemination and non-specific binding with other cells.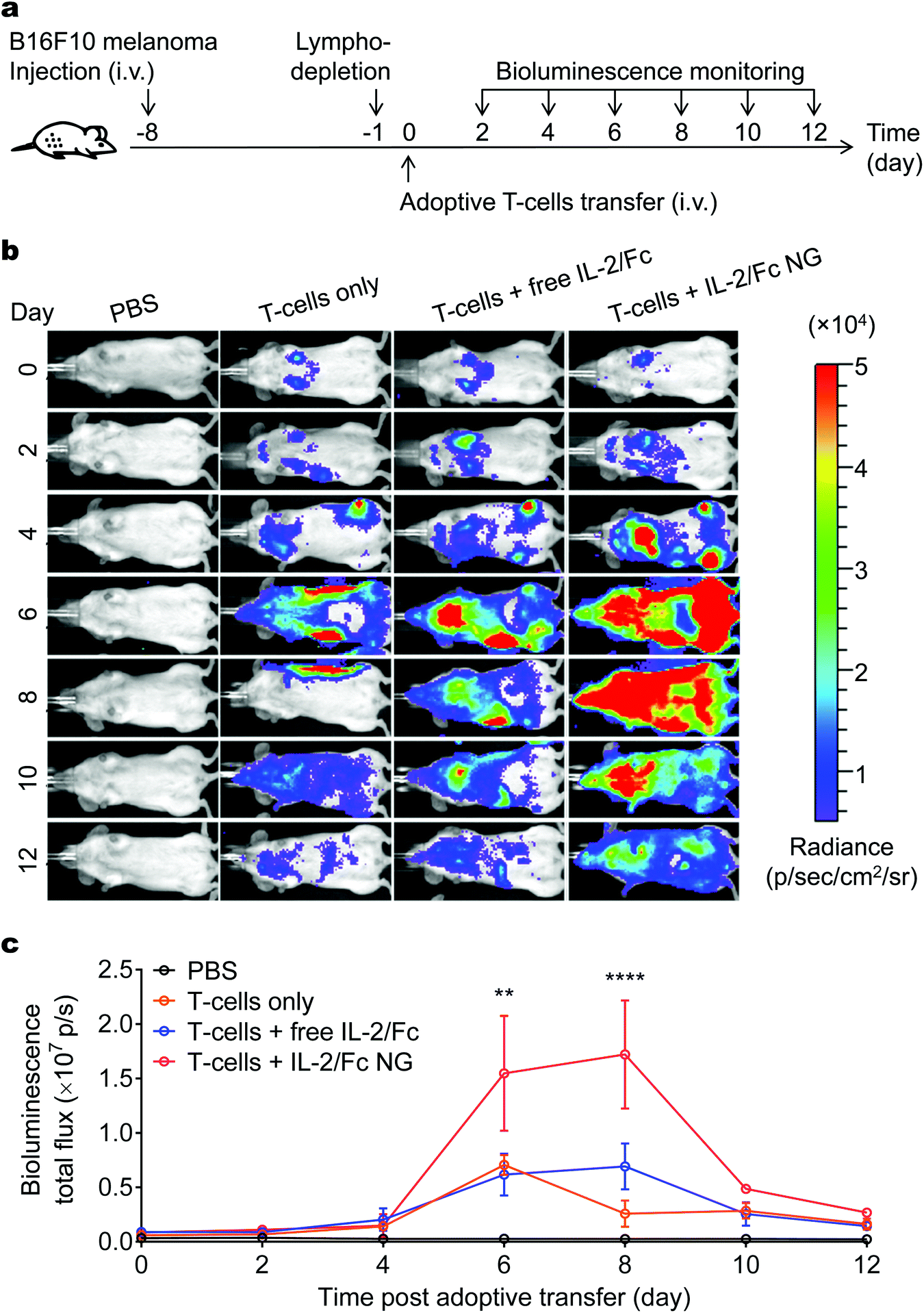 | ||
| Fig. 5 IL-2/Fc NG backpacks promote potent expansion of tumor-reactive T-cells in mice with pulmonary melanoma metastases. B16F10 melanoma cells (1.0 × 106) were i.v. injected into Thy1.2+ C57Bl/6 mice and allowed to establish pulmonary metastasis for 7 days. Mice were then sublethally lymphodepleted by irradiation at day-1, followed by i.v. infusion of activated pmel-1 luciferase-expressing Thy1.1+ CD8+ T-cells (1.0 × 107) at day 0. Mice received injections of PBS, T-cells only, T-cells followed by i.v. injection of free IL-2/Fc (60 μg), or T-cells backpacked with IL-2/Fc NGs at the same dose. In vivo bioluminescence imaging was performed every other days until day 12. (a) Experimental timeline. (b) In vivo bioluminescence imaging. (c) Quantification of bioluminescence signal from b. Data represent the mean ± s.e.m. (n = 4 per group). *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 by One-Way ANOVA and Tukey's tests. | ||
We further investigated the expansion of intratumoral T-cells in a mouse model with subcutaneous B16F10 melanoma tumors. The mice received the similar treatment of adoptive transfer of pmel-1 CD8+ T-cells that can be distinguished using Thy1.1 marker. The tumors were harvested at day 13 post ACT for flow cytometry analyses (Fig. 6a). There was a 80-fold greater expansion of transferred pmel-1 T-cells with surface-bound IL-2/Fc NGs in tumors than that with i.v. administered soluble IL-2/Fc at the equivalent dose (Fig. 6b and c). Importantly, systemic delivery of IL-2/Fc expanded endogenous regulatory T-cells (Tregs) in a non-specific way promoting the immunosuppression in the TME (Fig. 6d). By contrast, IL-2/Fc delivered by NG backpacks only expanded the transferred CD8+ T-cells but not the endogenous Tregs compared to T-cell only group (Fig. 6d). With upregulated expression of high affinity IL-2 receptor α (IL-2Rα), Tregs have higher sensitivity to IL-2 signaling compared to CD8+ T-cells.18 Soluble IL-2/Fc delivered systemically preferentially expanded Tregs likely due to the high ligand–receptor affinity, whereas IL-2/Fc delivered by NG backpacks focused the cytokine action on the transferred tumor-reactive T-cells without stimulating bystander cells. The highly selective expansion of tumor-reactive T-cells was thus achieved with IL-2/Fc NG backpacks for potentially enhanced safety and anti-tumor efficacy.
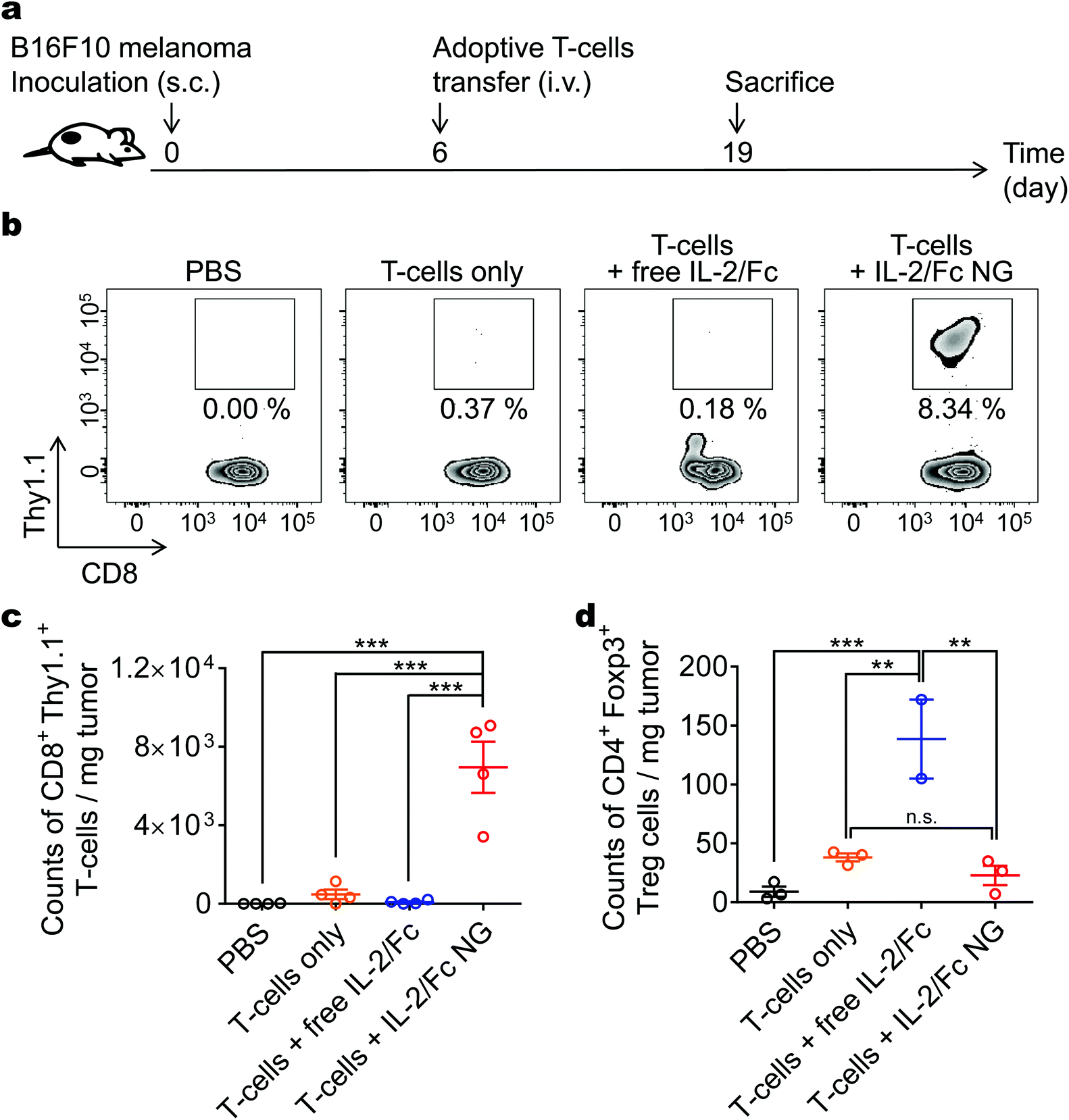 | ||
| Fig. 6 IL-2/Fc NG backpacks promote specific expansion of tumor-reactive T-cells in mice with subcutaneous melanoma. B16F10 melanoma cells (1.0 × 106) were s.c. inoculated on the flanks of Thy1.2+ C57Bl/6 mice and allowed to establish primary tumors for 6 days. Mice were then i.v. infused with activated Thy1.1+ pmel-1 CD8+ T-cells (1.0 × 107) at day 6. Mice received injections of PBS, T-cells only, T-cells followed by i.v. injection of free IL-2/Fc (80 μg), or T-cells backpacked with IL-2/Fc NGs at the same dose. Tumor-infiltrating lymphocytes were analyzed with flow cytometry at day 19 when mice were sacrificed. (a) Experimental timeline. (b) Representative flow cytometry plots showing the frequencies of tumor-infiltrating Thy1.1+ pmel-1 CD8+ T-cells among all the CD8+ T-cells. (c, d) Counts of tumor-infiltrating Thy1.1+ CD8+ T-cells (c) and endogenous CD4+ Foxp3+ regulatory T-cells (Tregs) (d). Data represent the mean ± s.e.m. (n = 3–4 per group). *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 by One-Way ANOVA and Tukey's tests; n.s., not significant. | ||
NG backpack delivery prevents the systemic toxicity of adjuvant cytokine and augments anti-metastasis efficacy of ACT
IL-2/Fc given systemically has been reported to be toxic to the treated mice.19–21 Here, we sought to evaluate whether NG delivery could diminish the toxicity of adjuvant cytokine in adoptive T-cell therapy. In a similar subcutaneous B16F10 melanoma model as that in Fig. 5, the mice were adoptively transferred with activated pmel-1 CD8+ T-cells in tandem with multiple doses of free IL-2/Fc or IL-2/Fc NG (Fig. 7a). Acute toxicity was observed in mice treated with T-cells with free IL-2/Fc injected i.v. as the body weight of mice dropped dramatically compared to the non-treated group (Fig. 7b). In contrast, there was no significant body weight change of mice treated with T-cells only or T-cells with IL-2/Fc NGs. This observation prompted us to further investigate the toxicity in vital tissues of the treated mice. We noticed the weight of excised spleens was 2.3-fold larger in mice treated with T-cells and free IL-2/Fc than that of mice in PBS control group (Fig. 7c) indicating severe systemic inflammation.22 This is consistent with previous reports that systemic IL-2 delivery led to rapid splenomegaly because of over-proliferation of endogenous lymphocytes.23,24 Further histological analyses revealed that the distinct demarcation of white and red pulps was lost in the spleens from free IL-2/Fc treated mice validating the intense lymphocyte expansion and the disorganization of splenic architecture (Fig. 7f). As a comparison, IL-2/Fc delivered by NG backpacks resulted in no spleen damage (Fig. 7c and f).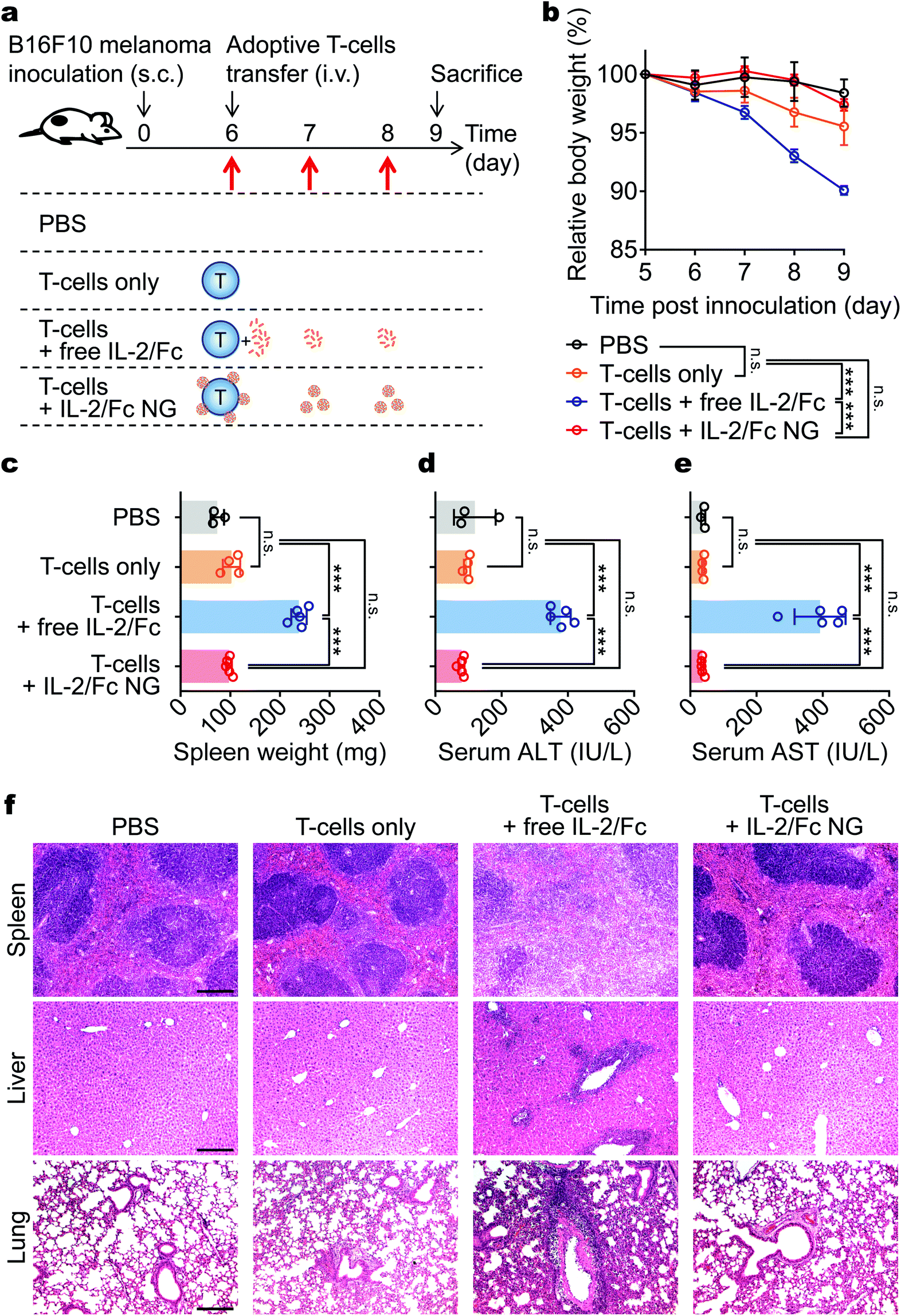 | ||
| Fig. 7 IL-2/Fc NG backpacks prevent the systemic toxicity of IL-2/Fc in mice. B16F10 melanoma cells (1 × 106) were s.c. inoculated on the flanks of Thy1.2+ C57Bl/6 mice and allowed to establish primary tumors for 6 days. At day 6, mice received i.v. injection of PBS, activated pmel-1 CD8+ T-cells (1.0 × 107) alone, or T-cells with free IL-2/Fc (daily i.v. injections of 60 μg each dose at day 6, 7, and 8), or IL-2/Fc NGs at equivalent dose (surface-bound NG backpacks at day 6; non-coupled NGs at day 7 and day 8 through i.v. injections). Serum biochemistry and histology were analyzed when mice were sacrificed at day 9. (a) Experimental timeline and groups. (b) Body weight, normalized to that at day 5 was monitored over time. (c) Spleen weights were measured following excision from mice. (d, e) Serum levels of ALT (d) and AST (e) were measured from samples collected at day 9. (f) Representative H&E-stained histological sections of spleen, liver and lung collected from the mice. Scale bar: 200 μm. Data represent the mean ± s.e.m. (n = 3–5 per group). *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 by One-Way ANOVA and Tukey's tests; n.s., not significant. | ||
The liver and lungs are vital organs preferentially infiltrated by transferred activated T-cells during the first few days post adoptive transfer.25,26 By measuring the serum levels of liver enzymes, we noticed markedly elevated levels of ALT and AST in the mice of free IL-2/Fc group but not in the other groups suggesting severe hepatotoxicity caused by systemic IL-2/Fc (Fig. 7d and e). In addition, massive portal inflammatory infiltration and parenchymal injury were observed in the livers from free IL-2/Fc treated mice (Fig. 7f). Similarly, the heavy inflammatory infiltration into the alveolar space together with large lesion areas were observed in lungs from mice receiving free IL-2/Fc treatment (Fig. 7f). In contrast, all the mice treated with T-cells with IL-2/Fc NG backpacks at an equivalent dose showed no overt toxicity likely because the delivery of IL-2/Fc by TCR-responsive NGs tethered on T-cell surface restricted the activity of IL-2/Fc in tumors minimizing the systemic exposure of free IL-2/Fc.
Next, we investigated whether the NG backpack delivery of adjuvant cytokine could enhance therapeutic efficacy of ACT. In a metastasis-mimicking mouse model, B16F10 melanoma cells were inoculated i.v. to induce lung tumor nodules (Fig. S6a†). Mice receiving the adoptive transfer of pmel-1 CD8+ T-cells backpacked with IL-2/Fc NGs exhibited markedly reduced number of lung metastatic nodules and lower grades of severity of metastasis burden compared to that of mice receiving T-cells only (Fig. S6b–e†).
Conclusions
In summary, we developed redox-responsive IL-2/Fc NGs to deliver adjuvant cytokines for enhanced efficacy and safety of adoptive T-cell therapy. Adoptively transferred tumor-reactive T-cells backpacked with IL-2/Fc NGs showed potent and specific expansion in vitro and in vivo. Intriguingly, controlled release of IL-2 from NGs in response to TCR signaling maintains a low effective concentration of the cytokine leading to preferential differentiation into CD8+ memory T-cells with reduced exhaustion. Compared to free soluble IL-2/Fc administered systemically, IL-2/Fc delivered by T-cell surface-bound NGs showed no overt toxicity even with repeating dosages and enhanced the anti-metastases efficacy in a mouse melanoma model. This chemical strategy for tissue and cell specific delivery of adjuvant cytokines in ACT therapy can be extended to other γ-chain cytokines, and other immunomodulatory agents such as immune-stimulant antibody agonists, checkpoint blockade antibodies, or small molecules. Genetic engineering approaches that are actively being pursued in the field have also achieved controlled cytokine secretion through activation of TCR signaling-regulated transcription factors.27,28 Compared to many genetic approaches that have shown elegant control of the activation and function of ACT T-cells,29–31 this synthetic approach has the unique advantages allowing for the delivery a wide variety of adjuvant molecules (including small molecules that cannot be recombinantly expressed by cells) and a pre-determined dosage in the surface-bound backpacks with minimum risk of runaway stimulation of transferred T-cells that could lead to severe toxicities.32,33Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by Swiss National Science Foundation (Project grant 315230_173243), the Fondation Pierre Mercier pour la science, ISREC Foundation with a donation from the Biltema Foundation, Novartis Foundation for medical-biological Research (17A058), the École polytechnique fédérale de Lausanne (EPFL), the Marble Center for Nanomedicine, and the NIH award (CA172164). DJI is an investigator of the Howard Hughes Medical Institute.References
- A. D. Fesnak, C. H. June and B. L. Levine, Nat. Rev. Cancer, 2016, 16, 566–581 CrossRef CAS PubMed.
- S. Han, O. Latchoumanin, G. Wu, G. Zhou, L. Hebbard, J. George and L. Qiao, Cancer Lett., 2017, 390, 188–200 CrossRef CAS PubMed.
- S. B. Stephan, A. M. Taber, I. Jileaeva, E. P. Pegues, C. L. Sentman and M. T. Stephan, Nat. Biotechnol., 2015, 33, 97–101 CrossRef CAS PubMed.
- K. Liu and S. A. Rosenberg, J. Immunol., 2001, 167, 6356–6365 CrossRef CAS.
- Y. Zheng, M. T. Stephan, S. A. Gai, W. Abraham, A. Shearer and D. J. Irvine, J. Controlled Release, 2013, 172, 426–435 CrossRef CAS PubMed.
- C. M. Paulos, C. Wrzesinski, A. Kaiser, C. S. Hinrichs, M. Chieppa, L. Cassard, D. C. Palmer, A. Boni, P. Muranski, Z. Yu, L. Gattinoni, P. A. Antony, S. A. Rosenberg and N. P. Restifo, J. Clin. Invest., 2007, 117, 2197–2204 CrossRef CAS PubMed.
- R. Andersen, M. Donia, E. Ellebaek, T. H. Borch, P. Kongsted, T. Z. Iversen, L. R. Hölmich, H. W. Hendel, Ö. Met, M. H. Andersen, P. T. Straten and I. M. Svane, Clin. Cancer Res., 2016, 22, 3734–3745 CrossRef CAS PubMed.
- R. Baluna and E. S. Vitetta, Immunopharmacology, 1997, 37, 117–132 CrossRef CAS.
- E. A. Grimm, A. Mazumder, H. Z. Zhang and S. A. Rosenberg, J. Exp. Med., 1982, 155, 1823–1841 CrossRef CAS.
- S. A. Rosenberg, J. Immunol., 2014, 192, 5451–5458 CrossRef CAS PubMed.
- G. C. Sim, N. Martin-Orozco, L. Jin, Y. Yang, S. Wu, E. Washington, D. Sanders, C. Lacey, Y. Wang, L. Vence, P. Hwu and L. Radvanyi, J. Clin. Invest., 2014, 124, 99–110 CrossRef CAS PubMed.
- V. Kalia, S. Sarkar, S. Subramaniam, W. N. Haining, K. A. Smith and R. Ahmed, Immunity, 2010, 32, 91–103 CrossRef CAS PubMed.
- L. Tang, Y. Zheng, M. B. Melo, L. Mabardi, A. P. Castaño, Y. Q. Xie, N. Li, S. B. Kudchodkar, H. C. Wong, E. K. Jeng, M. V. Maus and D. J. Irvine, Nat. Biotechnol., 2018, 36, 707–716 CAS.
- E. F. Zhu, S. A. Gai, C. F. Opel, B. H. Kwan, R. Surana, M. C. Mihm, M. J. Kauke, K. D. Moynihan, A. Angelini, R. T. Williams, M. T. Stephan, J. S. Kim, M. B. Yaffe, D. J. Irvine, L. M. Weiner, G. Dranoff and K. D. Wittrup, Cancer Cell, 2015, 27, 489–501 CrossRef CAS PubMed.
- K. M. Huster, V. Busch, M. Schiemann, K. Linkemann, K. M. Kerksiek, H. Wagner and D. H. Busch, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5610–5615 CrossRef CAS PubMed.
- T. Kaartinen, A. Luostarinen, P. Maliniemi, J. Keto, M. Arvas, H. Belt, J. Koponen, P. I. Mäkinen, A. Loskog, S. Mustjoki, K. Porkka, S. Ylä-Herttuala and M. Korhonen, Cytotherapy, 2017, 19, 1130 CrossRef PubMed.
- W. W. Overwijk, M. R. Theoret, S. E. Finkelstein, D. R. Surman, L. A. de Jong, F. A. Vyth-Dreese, T. A. Dellemijn, P. A. Antony, P. J. Spiess, D. C. Palmer, D. M. Heimann, C. A. Klebanoff, Z. Yu, L. N. Hwang, L. Feigenbaum, A. M. Kruisbeek, S. A. Rosenberg and N. P. Restifo, J. Exp. Med., 2003, 198, 569–580 CrossRef PubMed.
- S. G. Zheng, J. Wang, P. Wang, J. D. Gray and D. A. Horwitz, J. Immunol., 2007, 178, 2018–2027 CrossRef CAS.
- B. Kwong, S. A. Gai, J. Elkhader, K. D. Wittrup and D. J. Irvine, Cancer Res., 2013, 73, 1547–1558 CrossRef CAS PubMed.
- Y. Zhang, N. Li, H. Suh and D. J. Irvine, Nat. Commun., 2018, 9(6), 1–15 Search PubMed.
- J. T. Sockolosky, E. Trotta, G. Parisi, L. Picton, L. L. Su, A. C. Le, A. Chhabra, S. L. Silveria, B. M. George, I. C. King, M. R. Tiffany, K. Jude, L. V. Sibener, D. Baker, J. A. Shizuru, A. Ribas, J. A. Bluestone and K. C. Garcia, Science, 2018, 359, 1037–1042 CrossRef CAS PubMed.
- V. Bronte and M. J. Pittet, Immunity, 2013, 39, 806–818 CrossRef CAS PubMed.
- M. A. Pozniak, P. S. Christy, M. R. Albertini, S. M. Duffek and J. H. Schiller, Cancer, 1995, 75, 2723–2741 CrossRef.
- R. Moriggl, D. J. Topham, S. Teglund, V. Sexl, C. McKay, D. Wang, A. Hoffmeyer, J. Van Deursen, M. Y. Sangster, K. D. Bunting, G. C. Grosveld and J. N. Ihle, Immunity, 1999, 10, 249–259 CrossRef CAS PubMed.
- B. John and I. N. Crispe, J. Immunol., 2004, 172, 5222–5229 CrossRef CAS.
- E. Galkina, J. Thatte, V. Dabak, M. B. Williams, K. Ley and T. J. Braciale, J. Clin. Invest., 2005, 115, 3473–3483 CrossRef CAS PubMed.
- M. Chmielewski, C. Kopecky, A. A. Hombach and H. Abken, Cancer Res., 2011, 71, 5697–5706 CrossRef CAS PubMed.
- D. Chinnasamy, Z. Yu, S. P. Kerkar, L. Zhang, R. A. Morgan, N. P. Restifo and S. A. Rosenberg, Clin. Cancer Res., 2012, 18, 1672–1683 CrossRef CAS PubMed.
- C.-Y. Wu, K. T. Roybal, E. M. Puchner, J. Onuffer and W. A. Lim, Science, 2015, 350, aab4077–aab4077 CrossRef PubMed.
- C. C. Kloss, M. Condomines, M. Cartellieri, M. Bachmann and M. Sadelain, Nat. Biotechnol., 2013, 31, 71–75 CrossRef CAS PubMed.
- V. D. Fedorov, M. Themeli and M. Sadelain, Sci. Transl. Med., 2013, 5, 1–12 Search PubMed.
- R. A. Morgan, J. C. Yang, M. Kitano, M. E. Dudley, C. M. Laurencot and S. A. Rosenberg, Mol. Ther., 2010, 18, 843–851 CrossRef CAS PubMed.
- L. Zhang, R. A. Morgan, J. D. Beane, Z. Zheng, M. E. Dudley, S. H. Kassim, A. V. Nahvi, L. T. Ngo, R. M. Sherry, G. Q. Phan, M. S. Hughes, U. S. Kammula, S. A. Feldman, M. A. Toomey, S. P. Kerkar, N. P. Restifo, J. C. Yang and S. A. Rosenberg, Clin. Cancer Res., 2015, 21, 2278–2288 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8bm01556b |
| This journal is © The Royal Society of Chemistry 2019 |