
Surgery-free injectable macroscale biomaterials for local cancer immunotherapy
Kewen
Lei
 a and
Li
Tang
*ab
a and
Li
Tang
*ab
aInstitute of Materials Science & Engineering, École polytechnique fédérale de Lausanne (EPFL), Lausanne, Switzerland CH-1015
bInstitute of Bioengineering, École polytechnique fédérale de Lausanne (EPFL), Lausanne, Switzerland CH-1015. E-mail: li.tang@epfl.ch
First published on 28th December 2018
Abstract
Immunotherapy can harness the power of host's immune system to fight cancer. In the last few decades, tremendous progress has been made in this field, with remarkable clinical successes achieved consisting of a durable response in a fraction of patients. However, there are enormous challenges to extending this therapy to the majority of cancer patients while retaining minimal adverse effects. Local immunotherapy is a promising approach for concentrating immunomodulation in situ without systemic exposure, therefore minimizing systemic toxicities. More importantly, local immunomodulation can still lead to systemic effects that confer overall anticancer immunity to eradicate disseminated diseases. To facilitate these local immunotherapies, a wide range of biomaterials have been developed as delivery systems to protect the locally injected immune-related therapeutics and extend their retention. Surgery-free injectable macroscale biomaterials are one of the most promising classes of biomaterials developed to date, as they are suitable for minimally invasive injection with needles or catheters and form a biocompatible three-dimensional matrix in situ as a drug-depot for controlled local delivery. In this mini-review, we provide an overview of the recent advancements in applying injectable macroscale biomaterials in local cancer immunotherapy by highlighting some recent examples. We compare various injectable biomaterials with different gelation mechanisms and discuss their applications in the delivery of immunomodulators, immune cells, and cancer vaccines. We also discuss current challenges and provide a perspective for the future development of injectable macroscale biomaterials in cancer immunotherapy.
1. Introduction
Cancer immunotherapy, a treatment that harnesses the patient's own immune system to fight cancer, has recently shown dramatic clinical success.1 For example, checkpoint blockade therapies have been widely used to treat a variety of malignancies leading to unprecedented durable responses in a fraction of treated patients.2 Adoptive T-cell therapy, such as chimeric antigen receptor (CAR) T-cell therapy, has been effective in eradicating some hematopoietic cancers in the clinic.3 However, the percentage of cancer patients benefiting from immunotherapy remain modest. To realize its full clinical potential, strategies are being actively explored to further enhance anti-cancer immunity through combination therapies or using potent immunostimulant agents. However, balancing anti-cancer immunity and the concurrent immune-related toxicities remains a major challenge.4Local immunotherapy concentrates the immunomodulatory activity in situ while eliminating systemic exposure, therefore minimizing the adverse systemic effects.5 This local treatment could also lead to systemic immunity that eradicates disseminated diseases, a phenomenon called “abscopal effect”.6,7 To facilitate local immunotherapies, a wide range of biomaterials have been developed as delivery systems to protect the locally injected therapeutics and extend their retention.8–13 Among others, surgery-free injectable macroscale biomaterials are of particular interest, as these materials are suitable for injection with needles or catheters and form a biocompatible three-dimensional matrix in situ as a drug-depot for local therapeutic delivery (Fig. 1). This easy and minimally invasive administration without the need for surgical implantation is clinically applicable14,15 and thus particularly promising in the delivery of immunotherapies.
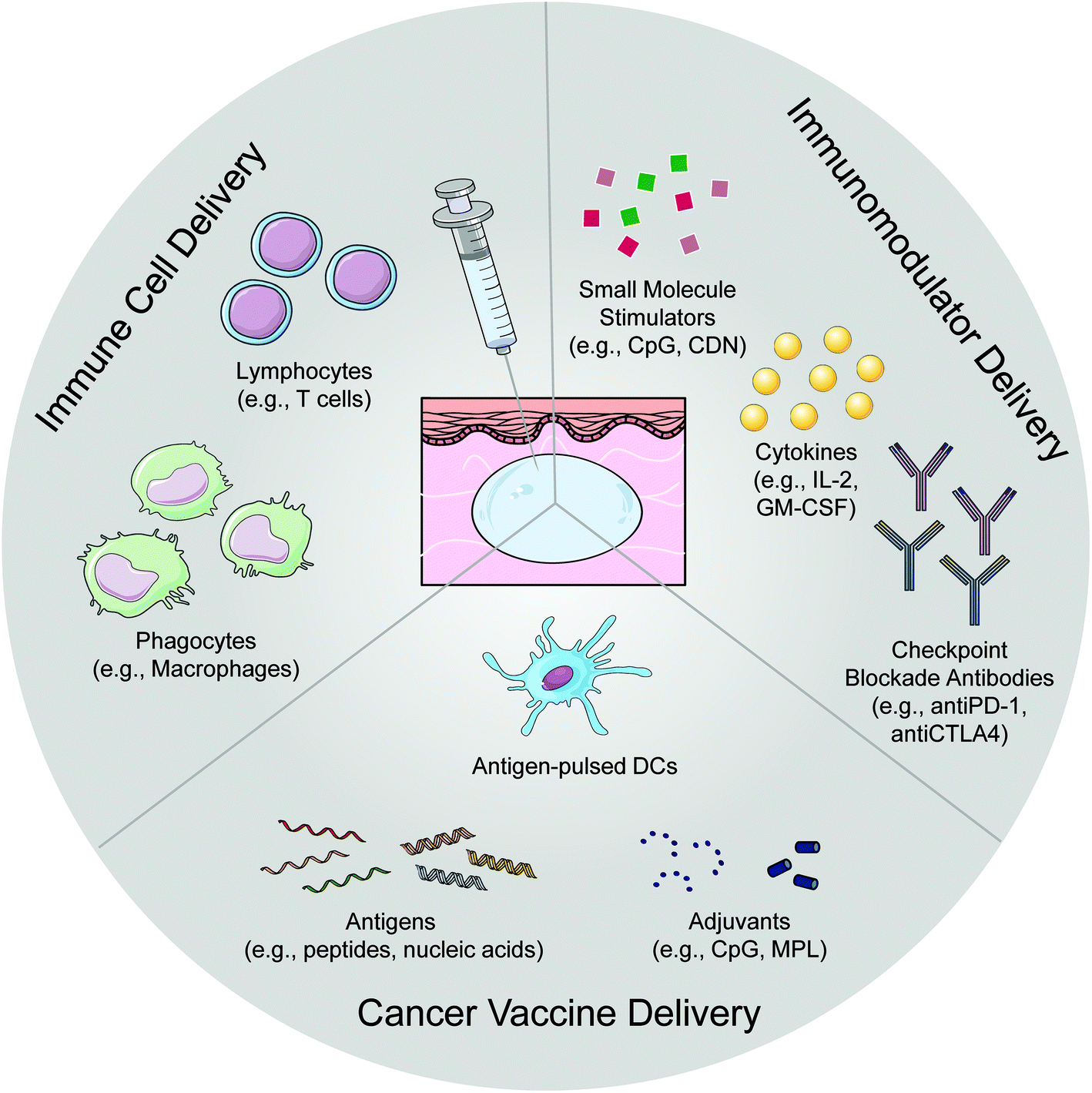 | ||
| Fig. 1 Schematic illustration of injectable macroscale biomaterials for the delivery of various cancer immunotherapies, including immunomodulators, cancer vaccines, and immune cells. Injectable macroscale biomaterials can be easily implanted into tissues via a simple injection and form a depot in situ for sustained and controlled therapeutic release. CpG, unmethylated cytosine–phosphate–guanine oligodeoxynucleotides; CDN, synthetic cyclic dinucleotide; IL-2, interleukin-2; GM-CSF, granulocyte–macrophage colony-stimulating factor; PD-1, programmed cell death protein 1; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; DCs, dendritic cells; MPL, monophosphoryl lipid A. | ||
Injectable macroscale biomaterials typically possess certain physical properties, such as a liquid form, a shear-thinning property or a high compression strain, which enable the injectability.16,17 A great number of these biomaterials have been developed with diverse physicochemical properties, including physically crosslinked hydrogels, chemically crosslinked hydrogels, cryogels, inorganic scaffolds, etc.18–21 Therapeutics with diverse physicochemical properties (e.g., positive charge, negative charge, hydrophilicity, hydrophobicity) can be loaded into those injectable macroscale biomaterials with a high encapsulation efficiency in defined doses. Biomaterials loaded with mono or combination therapies serve as reservoirs for controlled and sustained local release of encapsulated therapeutics upon injection, resulting in a concentrated local activity that minimizes systemic dissemination.
Other depot-forming macroscale biomaterials, such as implantable biomaterials, which are not injectable but require a surgery for implantation, can also be applied in immunotherapeutic delivery22,23 and have been reviewed previously.24,25 Nano- or micro-sized biomaterials that are injected locally without forming a macroscale matrix are not included in the discussion. In this mini-review, we give an overview of the recent developments in applications of injectable macroscale biomaterials in cancer immunotherapy delivery, including soluble immunomodulator delivery, immune cell delivery, and cancer vaccine delivery (Fig. 1). The features and applications of various injectable macroscale biomaterials are discussed by highlighting some recent advances in the field (Table 1). We also share our perspectives on the future directions for the development of these biomaterials for cancer immunotherapy.
| Biomaterial type | Composition | Cargo | Gelation factor | Tumor model | Ref. |
|---|---|---|---|---|---|
Abbreviations: —, not performed; Fmoc, fluorenylmethyloxycarbonyl; STING, stimulator of interferon genes; CpG, unmethylated cytosine–phosphate-guanine oligodeoxynucleotides; anti-PD-1, programmed cell death protein 1 antibody; P(Me-D-1MT), poly(L-methionine-co-dextro-1-methyl tryptophan); anti-PD-L1, programmed death-ligand 1 antibody; PEG, polyethylene glycol; PMNT, poly[4-(2,2,6,6-tetramethylpiperidine-N-oxyl) aminomethylstyrene]; PAAc, poly(acrylic acid); IL-12, interleukin-12; PVA, poly(vinyl alcohol); TSPBA, N1-(4-boronobenzyl)-N3-(4-boronophenyl)-N1,N1,N3,N3-tetramethylpropane-1,3-diaminium; GM-CSF, granulocyte-macrophage colony-stimulating factor; ICG, indocyanine green; DC, dendritic cell; OVA, ovalbumin; TCL, tumor cell lysates; Nap, naphthylacetyl; poly(I![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :C), polyinosinic:polycytidylic acid; GPDH, glycerol 2-phosphate disodium hydrate; TRP2180–188: tyrosinase-related protein 2 residues 180–188; ED-OVA, ethylenediamine-conjugated ovalbumin; mPEG-PLGA, methoxy-poly(ethylene glycol)-b-poly(lactic-co-glycolic acid); DC-LV-OVA, DC-targeting lentivirus expressing ovalbumin; MPL, monophosphoryl lipid A; PEI, polyethyleneimine; HRP, horseradish peroxidase; HP, hydroxyphenyl propionic acid; oAd, oncolytic adenovirus; PEGdA, polyethylene glycol diacrylate; Gel-PEG-Cys, thiolated poly(ethylene glycol)-grafted gelatin; TIL, tumor infiltrated lymphocytes. :C), polyinosinic:polycytidylic acid; GPDH, glycerol 2-phosphate disodium hydrate; TRP2180–188: tyrosinase-related protein 2 residues 180–188; ED-OVA, ethylenediamine-conjugated ovalbumin; mPEG-PLGA, methoxy-poly(ethylene glycol)-b-poly(lactic-co-glycolic acid); DC-LV-OVA, DC-targeting lentivirus expressing ovalbumin; MPL, monophosphoryl lipid A; PEI, polyethyleneimine; HRP, horseradish peroxidase; HP, hydroxyphenyl propionic acid; oAd, oncolytic adenovirus; PEGdA, polyethylene glycol diacrylate; Gel-PEG-Cys, thiolated poly(ethylene glycol)-grafted gelatin; TIL, tumor infiltrated lymphocytes. |
|||||
| 1. Immunomodulator delivery | |||||
| Supramolecular hydrogel | K2(SL)6K2 peptide | Synthetic STING agonist | Formation of β-sheet nanofiber | C57BL/6 mice with MOC2-E6E7 tumor | 59 |
| Supramolecular hydrogel | CpG-containing hexapod-like DNA | CpG | Formation of DNA double helix | C57BL/6J mice with EG7-OVA tumor | 61 |
| Supramolecular hydrogel | Alginate/Ca2+ | Anti-PD-1/celecoxib | Electrostatic interaction | C57BL/6 mice with B16F10 tumor or 4T1 lung metastases | 136 |
| Thermosensitive hydrogel | P(Me-D-1MT)-PEG-P(Me-D-1MT) triblock copolymer | Anti-PD-L1 | Micelle stacking via hydrophobic interaction | C57BL/6 mice with B16F10 tumor | 67 |
| Thermosensitive hydrogel | PMNT-PEG-PMNT and PAAc | IL-12 | Micelle stacking through hydrophobic and electrostatic interactions | Balb/c mice with C26 tumor | 68 |
| Chemically crosslinked injectable hydrogel | PVA and TSPBA | Anti-PD-L1/gemcitabine | Covalent bond formation between diol and boronic acid | C57BL/6 mice with B16F10 or 4T1 tumor | 72 |
| Injectable cryogel | Methacrylated gelatin | GM-CSF | Polymerization of acylate groups | — | 73 |
| 2. Immune cell delivery | |||||
| Chemically crosslinked injectable hydrogel | HP-modified gelatin/H2O2/HRP | DC and oAd-expressing IL-12 and GM-CSF | Enzymatic coupling of phenolic groups | C57BL/6 mice with LLC | 88 |
| Chemically crosslinked injectable hydrogel | PEGdA/Gel-PEG-Cys | M1 macrophage | Michael addition between acrylate group and sulfide | BALB/c nu/nu mice with MHCC97L xenograft | 87 |
| Thermosensitive hydrogel | Chitosan/NaHCO3/phosphate buffer | TIL or activated T cells | Hydrophobic and electrostatic interactions | — | 96 |
| 3. Cancer vaccine delivery | |||||
| Supramolecular hydrogel | Fmoc-KCRGDK peptide | JQ1- and ICG-loaded 4T1 tumor cells | Formation of self-assembled nanofiber | BALB/c mice with 4T1 or EMT6 tumor xenograft | 115 |
| Supramolecular hydrogel | Nap-modified D-tetrapeptide | OVA or X-ray-treated EG7 tumor cell | Formation of β-sheet nanofiber | C57BL/6J mice with B16-OVA or EG7 tumor | 109 |
| Supramolecular hydrogel | PEG-poly(L-valine) copolymer | TCL and poly(I:C) |
Physical aggregation of nanoassemblies via β-sheet and α-helix formations | C57BL/6 mice with B16 tumor | 117 |
| Supramolecular hydrogel | CpG-containing hexapod-like DNA | ED-OVA or R8FFRKSIINFEKL | Formation of DNA double helix | C57BL/6J mice with EG7-OVA tumor | 108 |
| Thermosensitive hydrogel | Chitosan/methylcellulose/GPDH | OVA or TRP2180–188/Quil A | Hydrophobic and electrostatic interactions | C57BL/6J mice with B16-OVA or B16 tumor | 126 |
| Thermosensitive hydrogel | mPEG-PLGA diblock copolymer | DC-LV-OVA/GM-CSF/MPL | Micelle stacking through hydrophobic interaction | C57BL/6J mice with B16-OVA tumor | 125 |
| Injectable cryogel | Methacrylated-alginate | Irradiated DD cell/GM-CSF/CpG | Polymerization of acylate groups | BALB/cJ mice challenged by DD breast cancer | 129 |
| Injectable inorganic scaffold | PEI-coated mesoporous silica microrod | Peptide antigens/GM-CSF/CpG | Stacking of high-aspect-ratio microrods | C57BL/6 mice with E7-TC-1, B16F10 or CT26 tumor | 133 |
| Injectable inorganic scaffold | Mesoporous silica microrod | OVA/GM-CSF/CpG | Stacking of high-aspect-ratio microrods | C57BL/6J mice with EG7-OVA tumor | 134 |
2. Injectable macroscale biomaterials
Many different types of injectable macroscale biomaterials have been developed to date. In general, these biomaterials can be categorized into (i) physically crosslinked injectable hydrogels, (ii) chemically crosslinked injectable hydrogels, (iii) injectable cryogels, and (iv) injectable inorganic scaffolds, the features of which are summarized in Table 2. In fact, injectable biomaterials have been widely applied in the clinic for various applications, such as cosmetic surgery,26 post-operation anti-adhesion,27 and tissue repairing.28 Many are available in the market including hyaluronic acid-based hydrogel for post-operation anti-adhesion (e.g., Adcon®, SprayShield®)29,30 and osteoarthritis treatment (e.g., Gel-One® and Euflexxa®).31 Recently, a collagen-based injectable scaffold (AUGMENT®)28 received the approval from USA-Food and Drug Administration (FDA) for bone regeneration, which is the first alternative to autograft in hindfoot and ankle arthrodesis.| Physically crosslinked injectable hydrogel | Chemically crosslinked injectable hydrogel | Injectable cryogel | Injectable inorganic scaffold | |
|---|---|---|---|---|
| Size | 1 mm– | 1 mm– | 1 mm–10 mm | 1 mm– |
| Backbone | Polymer or self-assembled structure | Polymer chain | Polymer chain or self-assembled structure | Inorganic microrod |
| Crosslinking structure | Physical aggregate | Covalent linkage | Covalent linkage or physical aggregate | Physical stacking |
| Porous structure | No | No | Yes | Yes |
| Mechanical strength | Low | Moderate to high | High | Moderate |
Physically crosslinked hydrogels are typically fabricated based on noncovalent interactions, such as hydrophobic interactions, hydrogen bonding, etc. They either remain non-crosslinked (as a solution) before injection or possess a shear-thinning property that enables their injectability. Among them, supramolecular hydrogels and thermosensitive hydrogels are two extensively studied systems. The former has a three-dimensional network that is stabilized by noncovalent interactions, such as hydrogen bond, π–π interactions, host–guest interactions, etc.32 As noncovalent interactions are reversible and weaker than covalent bonds under normal conditions, supramolecular hydrogels are intrinsically self-healing and can easily be injected into the target site via needle. These hydrogels can be made from diverse materials, including proteins, nucleic acids, polysaccharides, oligo- or poly-peptides, or synthetic polymers with pendant host and guest chemical groups (e.g., β-cyclodextrin and adamantane).33,34 This imparts huge structural and functional diversities to the supramolecular hydrogel system, which are beneficial for designing a custom-tailored delivery system for specific therapeutics. Particularly, programmable supramolecular hydrogels have attracted much attention recently as they can respond to complex biochemical stimuli orthogonally for precisely controlled release pattern of multiple cargos.35,36
Thermosensitive hydrogels are a special type of physical hydrogel whose gelation process is induced by temperature variation.37,38 It remains a solution before injection and quickly transforms into a gel at physiological temperatures. Basically, the building blocks of thermosensitive hydrogels feature an amphiphilic structure with segregated hydrophilic and hydrophobic domains. The temperature-induced gelation is usually driven by increased hydrophobic interactions upon elevated temperature that facilitate the formation of physical junctions of the three-dimensional network. Due to the nature of physical crosslinking, temperature-induced gelation is reversible. Also, the gelation temperature can be controlled by fine-tuning the hydrophilic/hydrophobic domain ratio.39 To date, various types of thermosensitive hydrogels have been developed, including polyethylene glycol (PEG)/polyester block copolymers, PEG/polypeptide block copolymers, amphiphilic poly(N-isopropylacrylamide) (PNIPAAm) copolymers, amphiphilic polyphosphazenes, chitosan derivatives, etc.37,38 Notably, various recent studies focused on the biomedical applications of PEG/polypeptide block copolymers due to their inherent biocompatibility and tunable secondary structure that well mimics the extracellular matrix microenvironment.40,41 Generally, the encapsulation conditions for thermosensitive hydrogels are typically mild, especially suitable for some environmentally sensitive drugs, such as proteins, because loading can be done in an aqueous system at room temperature or even at refrigerated temperature. Additionally, the temperature-induced physical gelling process largely maintains drug bioactivity as compared to chemically crosslinked injectable hydrogels that rely on chemical reactions for hydrogel network formation.42,43
Chemically crosslinked injectable hydrogels exploit covalent bonds to form the three-dimensional network. Compared to traditional chemically crosslinked hydrogels, these injectable crosslinking reactions occur at the target site in vivo surrounded by tissues,21 requiring the reactions being biocompatible without generating by-products toxic to normal tissues. Also, the reactions should be efficient to avoid excess reactive functional groups. To fulfill these requirements, some mild though highly efficient reactions can be used (e.g., click chemistry, disulfide formation, Michael addition, Schiff's base reaction, enzymatic reactions, and redox/photo-initiated polymerization).44 Chemically crosslinked injectable hydrogels typically have a higher mechanical strength and network stability than their physically crosslinked counterparts. These features are important in supporting cell/tissue ingrowth and fabricating defined or gradient functionalized patterns inside hydrogels or on hydrogel surfaces.45–47 Also, stimulus-responsive chemical bonds or enzymatically degradable moieties can be introduced into the hydrogel network to achieve responsive release of cargos or programmable degradation behaviors.48,49
Cryogel is a sponge-like network with interconnected micropores surrounded by a polymer wall. Generally, the microporous network is generated by freeze–thawing, and the matrix structure across the cryogel can be either chemically or physically crosslinked. Also, both natural and synthetic polymer can be used to construct cryogel.50 Due to the microporous structure and good interconnectivity, cryogel has advantages of quick diffusion of soluble factors and better cellular infiltration/ingrowth as compared to hydrogels without microporous structure.51 Although cryogel is a solid and usually possesses a high mechanical strength, it can be injectable if the cryogel is molded into a shape designed for passing through a needle. In recent years, nanocomposite-encapsulated injectable cryogels have emerged as an advanced technique for multi-functionality and improved performances in the delivery of therapeutics.52,53
Injectable inorganic scaffolds are a new form of macroscale biomaterial that use high-aspect-ratio inorganic microrods to generate the scaffold network. Different from physically or chemically crosslinked injectable hydrogels, these rely on the physical stacking of microrods to form a three-dimensional network structure and are injectable because of the microscale size of the building blocks that can pass through the needle. One unique characteristic of this system is that it has a highly rigid backbone made from microrod while retaining a micropore structure for cell/tissue ingrowth. It provides an alternative platform for the delivery of cell-based therapeutics or the recruitment of host cells in vivo.
3. Injectable macroscale biomaterials for immunomodulator delivery
Immunomodulators refer to any soluble factors that can directly control the immune response,54 such as cytokines, checkpoint blockade antibodies, co-stimulatory agonists, and chemical inhibitors. In cancer immunotherapy, immunomodulators stimulate the anti-tumor immune response. Systemic administration of those immunomodulators may lead to undesired immune stimulation and sometimes even lethal toxicity, especially in combination therapies.55,56 Therefore, a number of biomaterial-based strategies have been developed to improve immunomodulator targeting in tumor tissues to minimize this non-specific activation.57 Among different biomaterials, injectable macroscale biomaterials are a highly promising platform that can form a concentrated depot to confine the activity of immunomodulators to the target site without systemic dissemination. Here, we review the applications of injectable macroscale biomaterials in immunomodulator delivery by highlighting some recent examples.3.1. Physically crosslinked injectable hydrogels
DNA-based supramolecular hydrogels are also of great interest in immunomodulator delivery as they enable the precise network structure design and can intrinsically deliver DNA-based drugs, such as the toll-like receptor 9 agonist CpG,60,61 which was developed as a hydrogel in 2011 by Nishikawa et al.62 At that time, however, this system showed only marginal anti-tumor efficacy even in combination with a chemical anti-tumor drug doxorubicin. Recently, the same research group further combined the CpG-incorporated DNA-based hydrogel with photothermal therapy to enhance the immunostimulatory functions of CpG.61 In this system, a CpG-containing hexapod-like DNA structure was designed to crosslink the gold nanorod (AuNR), which functioned as a photosensitizer for photothermal therapy, modified with complementary oligo-DNAs on its surface. After mixing, they could form a shear-thinning hydrogel (designated as AuNR-CpG hydrogel) that could be injected by needle. Remarkably, the intratumoral injection of the AuNR-CpG hydrogel significantly inhibited the tumor growth in mice when combined with laser-induced photothermal therapy. Likely the hydrogel formulation plays an important role in generating robust immune responses via controlled immunomodulator release. As CpG is a commonly used adjuvant in vaccine studies,63,64 this system also has great potential in generating an effective cancer vaccine. In fact, some work has been done on this topic, and the details are discussed in the section “Injectable Macroscale Biomaterials for Cancer Vaccine Delivery”.
:polycytidylic acid (poly[I:C]).
 | ||
| Fig. 2 (A) Structure of P(Me-D-1MT)-PEG-P(Me-D-1MT) and ROS-triggered polymeric hydrophobicity transition. (B) Schematic illustration of localized hydrogel formation and bio-stimuli-triggered drug release and synergistic immunotherapy. (C) Average tumor volumes (n = 5), and (D) survival curves (n = 5) after a single treatment with various therapeutics (G1, PBS; G2, blank P[Me-D-1MT]-PEG-P[Me-D-1MT] hydrogel; G3, free D-1MT; and aPD-L1; G4, aPD-L1-loaded P[Me-D-1MT]-PEG-P[Me-D-1MT]) when the tumor volumes reached ∼110 mm3 on the seventh day (marked by red arrow). PEG, poly(ethylene glycol); D-1MT, dextro-1-methyl tryptophan; Me, L-methionine; P(Me-D-1MT), poly(L-methionine-co-dextro-1-methyl tryptophan); ROS, reactive oxygen species; DC, dendritic cell; aPD-L1, programmed death-ligand 1 antibody; IDO, indoleamine-pyrrole 2,3-dioxygenase; Treg, regulatory T cell. Adapted from ref. 67 with permission from Wiley-VCH. | ||
Another interesting thermosensitive hydrogel system is a composite consisting of a cationic polyamine–PEG–polyamine triblock copolymer with ROS-responsive side chains (nitroxide radical) and a anionic homopolymer poly(acrylic acid) for the sustained release of interleukin-12 (IL-12).68 Interestingly, the in vitro release patterns were similar among various protein cargos with different molecular weights and isoelectric points (e.g., bovine serum albumin, insulin, glucose oxidase, avidin, and neutravidin). Thus, it may provide a universal platform for the synchronized and sustained release of multiple protein cargos in one formulation. However, as the hydrogel-formulated IL-12 displayed only slightly increased anti-tumor efficacy, the release kinetic needs to be further optimized for maximum effect in the follow-up studies. Nevertheless, the application of this polyion-complex-based hydrogel system can potentially be extended for the delivery of various positively or negatively charged immunomodulators, as it has the potential to achieve high loading efficiency and controlled cargo release via electrostatic interaction between the hydrogel network and immunomodulators.
3.2. Chemically crosslinked injectable hydrogels
Despite the advantages of chemically crosslinked injectable hydrogels (e.g., persistent network structure for stable release kinetics), few studies to date have reported using them for immunomodulator delivery.71,72 One recent example is a ROS-responsive system developed by the Gu group for the co-delivery of a checkpoint inhibitor and a chemical anti-tumor drug gemcitabine (GEM) (Fig. 3).72 The hydrogel system was made of poly(vinylalcohol) (PVA) and N1-(4-boronobenzyl)-N3-(4-boronophenyl)-N1,N1,N3,N3-tetramethylpropane-1,3-diaminium (TSPBA). In this system, the TSPBA crosslinked the diols on PVA by complexing them with its two phenylboronic acid groups. The TSPBA could also be oxidized and scavenged upon elevated ROS levels, a key feature in the tumor microenvironment, to achieve tumor-site-specific release. As the crosslinking reaction occurs quickly, the PVA and TSPBA need to be loaded into a dual syringe separately before injection and mixed together only when they pass through the needle. Although this method ensures the injectability of the hydrogel system, it is worth noting that this method may generate heterogeneous regions with various physicochemical properties inside hydrogel, as the mixing process in the needle may not be sufficient. In in vivo studies, the peritumoral injection of the GEM and anti-PD-L1-encapsulated hydrogel system significantly inhibited tumor growth and prolonged survival in both B16F10 (Fig. 3) and low immunogenic 4T1 carcinoma models. Particularly in the B16F10 tumor model, the hydrogel system induced a systemic immune response that delayed the growth of distant tumor, as well, indicating that the hydrogel depot could also potentiate the efficacy of a chemo- and immuno-combination therapy. Interestingly, loss of tumor-promoting M2-like macrophage was observed in mice treated only with the blank hydrogel, implying that the ROS-scavenging property of the hydrogel system could remodel the tumor microenvironment for better anti-tumor efficacy. Therefore, the ROS scavenging moiety may be beneficial when designing injectable macroscale biomaterials for delivering macrophage-dependent immunomodulators. Finally, despite the injection method, this hydrogel system shows potential for clinical application, as it involves only two simple components: a small molecule crosslinker and PVA, which has been used as a drug additive in many clinical therapeutics.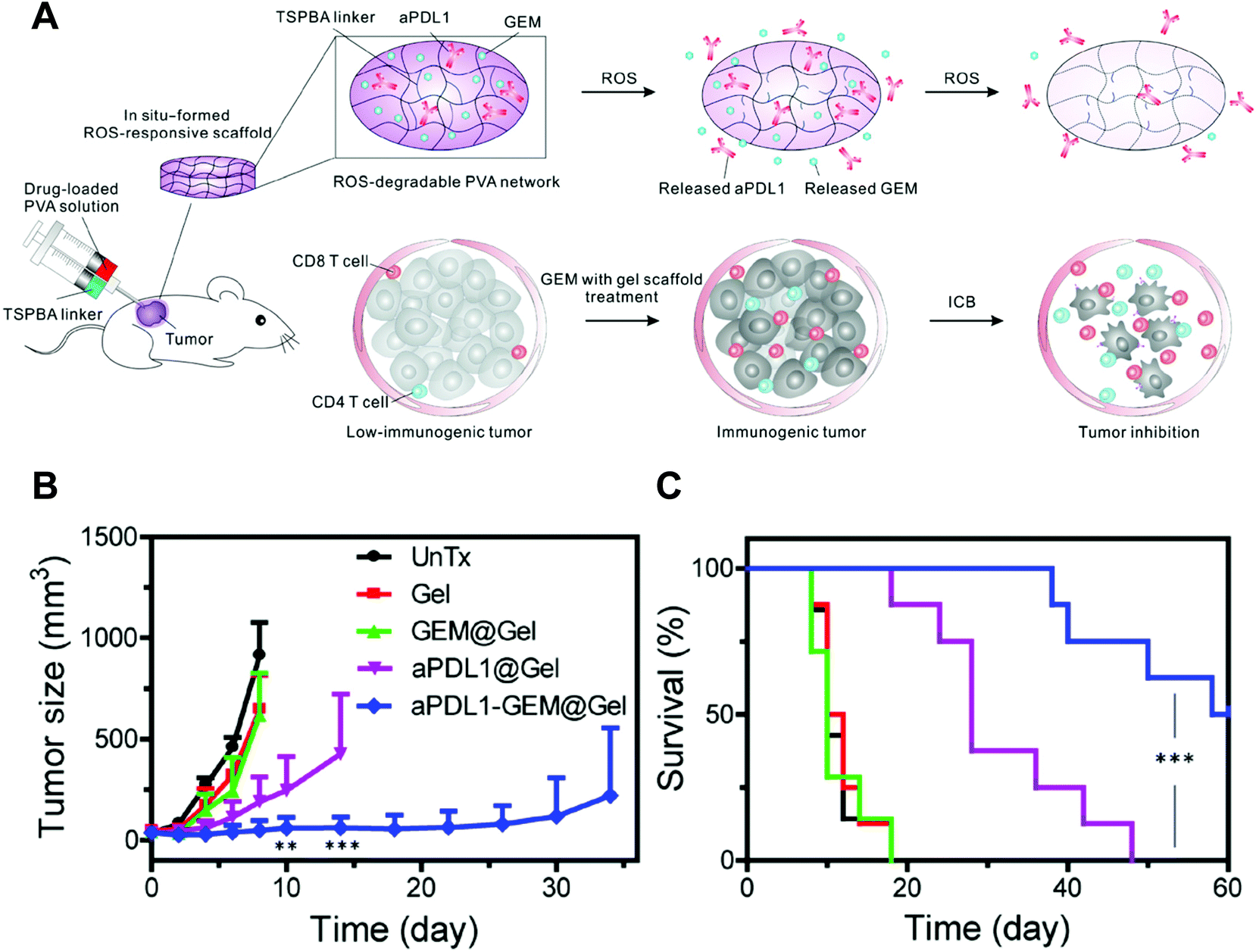 | ||
| Fig. 3 (A) Schematic of combination chemoimmunotherapy using a ROS-degradable hydrogel scaffold to deliver GEM and aPDL1 into the tumor microenvironment. (B and C) Local gel scaffold for inhibiting B16F10 melanoma growth in vivo. (B) Average tumor growth kinetics in control and treated groups (treatment started at day 0). Growth curves represent mean ± SEM; growth curves were stopped when the first animal of the corresponding group died. (C) Survival curves for the treated and control mice (n = 7 to 10). ROS, reactive oxygen species; PVA, poly(vinyl alcohol); TSPBA, N1-(4-boronobenzyl)-N3-(4-boronophenyl)-N1,N1,N3,N3-tetramethylpropane-1,3-diaminium; aPDL1, programmed death-ligand 1 antibody; GEM, gemcitabine; ICB, immune-checkpoint blockade; UnTx, untreated. Adapted from ref. 72 with permission from American Association for the Advancement of Science. | ||
3.3. Injectable cryogels
Compared to physically or chemically crosslinked injectable hydrogels, injectable cryogel possesses a unique microporous structure that enables cell entrance, meaning it can concentrate target cells in the depot for a more efficient drug action kinetic. The pioneering work in the delivery of immunomodulators using injectable cryogel was done by the Mooney group73 wherein methacrylated gelatin (GelMA) was used to form the cryogel polymer network via redox-initiated radical polymerization. The cryogel was preformed into a cylindrical shape (5 mm diameter, 2 mm thickness) for injection via needle. Interestingly, the cylindrical cryogel could pass through a 16G needle (inner diameter ∼1.2 mm) and recover its original shape after injection due to its high compression strain and elasticity. The immunomodulator granulocyte–macrophage colony-stimulating factor (GM-CSF) that attracts and stimulates immune cells (e.g., dendritic cells [DCs] and macrophages) could be loaded into the cryogel before crosslinking. After subcutaneous injection of the GM-CSF-loaded cryogel, it exhibited 20-fold higher live-cell recruitment into the matrix when compared to blank cryogel, which is of great interest in generating a local immunization depot. However, the in vitro release of GM-CSF from the cryogel showed a large initial burst of around 50%, so there is still room for improvement in prolonged cargo release for the sustained recruitment of host immune cells.4. Injectable macroscale biomaterials for immune cell delivery
In cancer immunotherapy, the ultimate anti-tumor responses are typically exerted by effector immune cells, such as cytotoxic CD8+ T cells, effector CD4+ T cells, and NK cells. Adoptive cell therapy has been developed into clinical applications including two CAR T-cell products recently approved by the FDA (Kymriah® and Yescarta®).74,75 Despite the exciting clinical results in the treatment of liquid cancers, the efficacy of adoptive cell immunotherapy in solid tumors remains modest due to the impaired infiltration of immune cells into solid tumors and the immuno-suppressive tumor microenvironment.76,77 Those cell-based immunotherapies could also induce serious or even fatal side effects.78–80To address these challenges, biomaterial-based strategies have been explored for the local delivery of therapeutic immune cells because they can enrich the immune cells inside tumor tissues, reverse the immunosuppressive tumor microenvironment, and alleviate systemic toxicities, such as cytokine release syndrome.81 Injectable macroscale biomaterials provide significant advantages in this particular application as they permit the simple mixing of therapeutic immune cells and injectable biomaterials ex vivo followed by the minimally invasive injection without surgery. In recent years, injectable hydrogel systems have been extensively investigated for the delivery of immune cells. For example, injectable alginate hydrogels were explored in a pioneer study by the Irvine group for the co-delivery of antigen-pulsed DC cells with supporting immunomodulators (e.g., CCL19, CCL21, and IL-15SA) for cancer immunotherapy.82,83 The DCs and immunomodulator-encapsulated hydrogel system outperformed free DCs plus immunomodulators in tumor growth inhibition and prolonging animal survival. Other examples are highlighted and discussed in this section.
4.1. Chemically crosslinked injectable hydrogels
Chemically crosslinked injectable hydrogels are favored for generating a stable depot with a defined mechanical strength and network structure for desired cell growth/differentiation. For example, several of these systems have been designed to deliver chondrocytes for cartilage tissue engineering.84–86 Recently, some have also been used to encapsulate immune cells for enhanced cancer immunotherapy.87,88 One example of this is a hydroxyphenyl propionic acid-conjugated gelatin (GHPA)-based hydrogel for the co-delivery of DCs and oncolytic adenovirus (oAd) that could co-express IL-12 and GM-CSF in tumor tissue.88 The in situ crosslinking of the hydrogel was initiated using the horseradish peroxidase (HRP) and H2O2 enzymatic reaction, which is highly efficient and biocompatible with living tissues, to generate multiple phenolic radicals on one single polymer chain that couple together to form a crosslinked network. Interestingly, the in vitro release of DCs from the hydrogel system was dependent on the stiffness, with a faster release rate coming from hydrogels with a lower stiffness. This phenomenon may be partly attributed to the lower crosslinking density of a hydrogel system with a lower stiffness, and it provides a possibility to control the DC-release kinetic for optimal immune responses. The hydrogel system could also help the accumulation of DCs and oAd in both tumor tissue and draining lymph nodes for a prolonged period of time. Importantly, a single intratumoral injection of a DC- and oAd-loaded hydrogel would facilitate greater infiltration of CD8+ T cells and induce a more potent antitumoral effect compared to that of free DCs plus oAd. Thus, the prolonged release of the immune cells has a positive impact on the outcome of immune-cell-based cancer treatment.Chemically crosslinked injectable hydrogels have also been used to deliver macrophages to tumor sites for enhanced cancer immunotherapy using a hydrogel system consisting of thiolated poly(ethylene glycol)-grafted gelatin (Gel-PEG-Cys) and poly(ethylene glycol) diacrylate (PEGdA).87 When mixing the two components, a gentle crosslinking occurred via the Michael addition between the thiol groups on Gel-PEG-Cys and the acrylic groups on PEGdA. The encapsulated M1-like macrophages retained their phenotype and displayed a similar cytokine-release profile to that of M1-like macrophages cultured in normal medium. The peritumoral injection of the M1-macrophage-encapsulated hydrogel significantly delayed the tumor growth in a xenograft model compared to a blank hydrogel group. It is worth noting that a treatment group with M1 macrophage alone was absent in the in vivo study, so it is unclear if the hydrogel system could promote the efficacy of macrophage-based cancer immunotherapy. In the tumor microenvironment, macrophages are polarized to an M2-like phenotype for tumor progression,89–91 so for future studies it will be of great interest to see if the injectable hydrogel system protects the macrophages from this polarization in the tumor microenvironment. Also, this system has the potential to generate an injectable hydrogel that can reprogram the tumor-promoting M2-like macrophage into the tumor-inhibiting M1-like macrophage phenotype.
4.2. Thermosensitive hydrogels
Thermosensitive hydrogels are a good candidate for immune-cell delivery as they can encapsulate cells in solution at a low temperature without cell damage. Additionally, no chemical additive is needed in the hydrogel system, so it has a better biocompatibility when compared with the chemically crosslinked hydrogel. These hydrogels have served as promising platforms for the delivery of stem cells and the control of their differentiation.92,93 In recent years, thermosensitive hydrogels have also emerged as a potential platform for the delivery of immune cells in cancer immunotherapy.94–96 A representative work is a thermosensitive chitosan hydrogel for the delivery of tumor-infiltrating lymphocytes and activated CD8+ T cells (Fig. 4).96 Initially, the chitosan was premixed with a phosphate-based buffer to generate the thermogelling system. The gelation time and mechanical strength of the hydrogel system could be finely tuned by varying the concentration of sodium bicarbonate in the buffer, and this provided a good platform for designing suitable physical features for immune cell proliferation. In the in vitro T-cell expansion experiment, it was found that the hydrogel system with a larger pore size and higher stiffness promoted a higher cell viability and proliferation rate of the encapsulated T cells (Fig. 4), which the authors suspected was due to the heightened gas diffusion and nutrient exchange inside the hydrogel. To corroborate this, some studies have shown that T cell behaviors are stiffness-dependent,97,98 with a higher stiffness potentially contributing to better in vitro T cell expansion. Intriguingly, CD8+ T cells tended to grow faster than CD4+ T cells in the thermosensitive chitosan hydrogel system (Fig. 4), but the related mechanism wasn't revealed in the article. It provides a hint that the type of immune response can be selectively controlled by using certain biomaterials. Finally, a primary Transwell T cell killing assay demonstrated that the encapsulated T cells could migrate from the thermosensitive chitosan hydrogel into the lower chamber seeded with tumor cells and perform normal, specific killing of these tumor cells. It suggested that the T cells delivered using thermosensitive chitosan hydrogels are likely to retain their potential for the specific killing of tumor cells in vivo. | ||
| Fig. 4 (A) Method of cell encapsulation and culture into chitosan-based biocompatible thermogels (CTGels). (B) Logarithmic scale of average cell numbers obtained at different time points from the supernatant and from CTGels 1 or 2. The storage modulus of CTGels 1 and 2 were ∼2.4 kPa and 3.5 kPa, respectively. (C) Graph of average percent of CD3+ cells that are also CD8+ or CD4+ was determined from cells collected from the media or those extracted from CTGel2 over a 15-day time course. Adapted from ref. 96 with permission from Elsevier. | ||
Other thermosensitive hydrogels, such as PEG-grafted chitosan and PEG-b-P(MEO2MA-co-OEGMA-co-NHSMA) copolymer, have also been explored for the encapsulation of T cells,94,95 though none of these thermosensitive hydrogel systems have been studied in vivo to determine their anti-tumor efficacy. In the future, the therapeutic effect of immune cell-encapsulated thermosensitive hydrogels needs to be demonstrated to better understand the in vivo behaviors of the encapsulated immune cells. It will be of great value in designing desirable thermosensitive hydrogel systems for the efficient delivery of immune cells and the precise modulation of anti-tumor immune responses.
5. Injectable macroscale biomaterials for cancer vaccine delivery
Cancer vaccines activate the host immune system against tumor antigens, either for prophylactic or therapeutic purposes. To generate a potent cancer vaccine, different strategies have been developed, including antigen-pulsed DCs, peptide subunit vaccine, antigen-encoding mRNA/DNA, and tumor cell lysates.99 It remains challenging, however, to efficiently deliver tumor antigens to the desired immune organs or cells.100 Subunit cancer vaccines, such as mRNA/DNA and peptide-based cancer vaccines, accumulate poorly in draining lymph nodes upon parental injections and rapidly disseminate into the systemic circulation.101,102 In recent years, a great deal of biomaterial-based cancer vaccine platforms, including injectable macroscale biomaterials, have emerged for improving the delivery efficiency and potency of cancer vaccines.103–106 Injectable macroscale biomaterials can serve as a depot for both antigens and adjuvants for slow release and extended retention in lymph nodes. Such a depot could also be designed as an artificial immune organ to recruit antigen-presenting cells (e.g., DCs) for enhanced antigen uptake and presentation. The application of those minimally invasive biomaterials in cancer vaccine delivery is reviewed here.5.1. Physically crosslinked injectable hydrogels
Peptide-based supramolecular hydrogels can form various self-assembled structures, such as nanofibers containing α-helixes or β-sheets, and self-assembling peptides could serve as adjuvants when conjugated with peptide or protein antigens.118–121 Various preclinical vaccines have been constructed based on these hydrogels against infectious diseases.122,123 Recently, some peptide-based supramolecular hydrogels have also been developed to elicit both effective humoral and cellular immune responses for cancer vaccines.109,111,114,116 A representative work is a system based on the naphthylacetic acid-modified tetra-peptide (Nap-GFFY; designated as L-gel),109 which possesses shear-thinning properties that enabled the injection and loading of the antigens through gentle shaking (Fig. 5). Intriguingly, the enantiomeric counterpart of the tetra-peptide (Nap-GDFDFDY; designated as D-gel) induced stronger humoral and cellular immune responses than L-gel, possibly due to better stability of D-gel in vivo. Particularly, the OVA-loaded D-gel better inhibited tumor growth than the monophosphoryl lipid A + OVA bolus formulation. Moreover, the potential of D-gel for fabricating whole-cell cancer vaccines was also tested. D-gel loaded with X-ray-treated EG7 tumor cell (XTC) significantly inhibited tumor growth (Fig. 5). However, the XTC-loading L-gel displayed a marginal anti-tumor efficacy similar as that of XTC alone. This suggests that the chemical composition of the hydrogel system, including isomeric effect, plays a key role in triggering potent anti-tumoral immune responses and should be considered for developing effective cancer vaccines.
 | ||
| Fig. 5 (A) Chemical structure of short peptide-based hydrogelators. The hydrogels could be converted to viscous solutions by shaking or vortexing, allowing the incorporation of antigens by simple mixing. (B and C) D-gel enhances the immunogenicity and therapeutic efficacy of X-ray-treated whole-cell vaccines. (B) Tumor growth in C57BL/6J mice (n = 6 per group) vaccinated with PBS, XTC, L-gel, D-gel, L-gel-XTC, or D-gel-XTC, and then challenged with EG7 tumor cells. Mice were vaccinated and inoculated as described above, and the tumor volume was monitored every three days. (C) The survival time of mice vaccinated with PBS, XTC, L-gel, D-gel, L-gel-XTC, or D-gel-XTC, respectively (n = 6 per group). Day 0 on the survival curves represents the day of tumor challenge. Nap, naphthylacetic acid; G, L-glycine; F, L-phenylalanine; Y, L-tyrosine; GD, D-glycine; FD, D-phenylalanine; XTC, X-ray-treated whole tumor cell; L-gel, hydrogel self-assembled from Nap-GFFY; D-gel, hydrogel self-assembled from Nap-GDFDFDY. Adapted from ref. 109 with permission from Wiley-VCH. | ||
Another interesting peptide-based supramolecular hydrogel system is a self-assembling and tumor-penetrable peptide Fmoc-KCRGDK (FK) for the construction of whole-cell cancer vaccines.115 The tumor cells were loaded with a PD-L1 expression inhibitor and a photoabsorbent indocyanine green (ICG). In this system, the potent anti-tumor immune response could be temporally triggered with laser radiation, providing a promising approach for precisely controlling anti-tumor responses using an external trigger.
So far, none of the recent injectable peptide-based supramolecular hydrogels have been explored for the delivery of subunit peptide antigens, which will be of great interest in the future for cancer vaccines. These peptide antigens can be chemically incorporated into the hydrogel network for prolonged presentation and precise control over the ratio between different peptide epitopes.
In recent years, the thermosensitive hydrogel systems for cancer vaccine fabrication have been almost entirely based on PEG/polyester block copolymers or chitosan.124–128 The amphiphilic PEG/polyester block copolymer is first assembled into micelles and then aggregated to form a hydrogel network upon increased temperature.19,39 As this hydrogel system intrinsically contains a large proportion of hydrophobic domains, it has an edge in the sustained release of hydrophobic drugs. In a recent study, the DC-recruiting factor GM-CSF was encapsulated in a methoxy-poly(ethylene glycol)-b-poly(lactic-co-glycolic acid) (mPEG-PLGA)-based thermosensitive hydrogel, and a sustained release manner of GM-CSF was achieved without a significant initial burst.125 As a result, the GM-CSF-loaded mPEG-PLGA thermosensitive hydrogel effectively enriched for DCs and macrophages in the depot after subcutaneous injection. After two immunization doses of DC-targeted and OVA-encoded lentiviral vectors as the antigen and monophosphoryl lipid A as the adjuvant, this system significantly suppressed tumor growth as compared to the system lacking GM-CSF. This indicated that DC enrichment in the hydrogel depot is crucial for enhanced vaccination efficacy. Interestingly, the authors injected the antigen/adjuvant mixture separately after the injection of GM-CSF-loaded hydrogel for the immunization. If the separated administration of antigen and adjuvant improves the vaccination efficacy, it may provide a better administration strategy for injectable macroscale biomaterial-based cancer vaccines.
The chitosan thermosensitive hydrogel is usually obtained by adding polyol salts (e.g., glycerol 2-phosphate disodium hydrate [GPDH]) into a chitosan solution. In one representative study, a chitosan/methylcellulose/GPDH ternary thermosensitive hydrogel was used to fabricate a subunit peptide cancer vaccine.127 The subcutaneous vaccination with TRP2 peptide180–188 (tyrosinase-related protein 2 residues 180–188; a B16 endogenous antigen) and Quil A (an adjuvant)-loaded chitosan thermosensitive hydrogel showed enhanced protection against tumor growth compared to TRP2 peptide180–188-pulsed DCs. Using OVA as a model antigen, it was further demonstrated that chitosan thermosensitive hydrogel-assisted vaccination could elicit stronger antigen-specific cytotoxic and memory CD8+ T-cell responses than DC-based vaccinations, suggesting that the sustained release of antigen and adjuvant could achieve superior efficacy compared to the antigen-pulsed DC strategy. For future studies, it will be of great interest to investigate whether the enhanced vaccination efficacy is a result of local DC activation at the hydrogel depot or enhanced accumulation of antigen and adjuvant in draining lymph nodes.
5.2. Injectable cryogels
Because cryogel intrinsically possesses a microporous structure and high network interconnectivity, it is ideal for generating a favorable artificial niche for the recruitment and activation of DCs. Recently, the Mooney group explored the feasibility of using injectable cryogels for fabricating whole-cell cancer vaccines.129,130 These vaccines typically use irradiated tumor cells to elicit a broad spectrum of immune responses to tumor-derived antigens, including tumor-associated antigens and neoantigens.131 In some clinical trials, genetically modified tumor cells expressing GM-CSF were used for vaccinations, but this method is costly and phase 3 trial results were disappointing, possibly due to the lack of efficient DC co-stimulation.132 Here, the cryogel is expected to provide a platform for the enrichment and durable stimulation of DCs. In Mooney's lab, a methacrylated alginate was used to fabricate the injectable cryogel sponge crosslinked via redox-initiated radical polymerization.130 The DC-stimulator GM-CSF and a CpG adjuvant could be physically incorporated into the system before crosslinking while the irradiated tumor cells were seeded on the fabricated cryogel sponge to serve as an antigen source. Strikingly, in a highly aggressive B16F10 tumor model, the cryogel-based whole-cell vaccine completely eradicated the established tumor in 40% of mice after subcutaneous immunization with only a second boosting. More recently, a similar methacrylated alginate-based injectable cryogel was also examined to generate a prophylactic whole-cell cancer vaccine, and its subcutaneous injection prevented tumor growth in up to 80% of mice.129Thus, this injectable cryogel system is a promising universal platform for fabricating effective whole-cell vaccines. However, the common problem in the existing systems is that the encapsulated cytokine or adjuvant (e.g., GM-CSF and CpG) displays a huge initial burst release (>50%), which is not ideal for maintaining a cytokine concentration gradient and a sufficient dose of adjuvants for effective DC recruitment and activation. A possible solution to this is to incorporate the cytokine or adjuvant-encapsulated polymer microspheres (e.g., poly[lactic-co-glycolic acid] and alginate microspheres) into the cryogel for sustained release of the cytokine and adjuvant.
5.3. Injectable inorganic scaffolds
At present, the vast majority of injectable macroscale biomaterial platforms rely on organic compounds such as synthetic polymers, peptides, and DNA for constructing the biomaterial network. Very few studies have focused on developing injectable inorganic biomaterials for the delivery of cancer immunotherapy and cancer vaccines. Until recently, the Mooney group has pursued the pioneering work on developing these scaffolds for effective cancer vaccines for both tumor-associated antigens and neoantigens.133,134 The building block of their injectable inorganic scaffold was a mesoporous silica microrod (MSR) with a high aspect ratio that could spontaneously assemble into a 3D scaffold in vivo (Fig. 6). This system has the advantage that a simple fabrication combines both a mesoporous structure for drug release and a microporous structure for cell recruitment. Notably, cell recruitment into the MSR scaffold was highly dependent on the structural parameters of the MSR, with the high-aspect-ratio MSR recruiting more DCs than the one formed from a low-aspect-ratio MSR. Moreover, both the mesopores on the MSR and the micropores between the stacking MSR were crucial for efficient cell recruitment, indicating that the physical features are important in designing injectable macroscale biomaterials to generate an effective immunization depot. Interestingly, a single immunization with GM-CSF, CpG, and OVA-loaded MSR scaffold (designated as MSR vaccine) induced both strong Th1 and Th2 responses, while the bolus form containing the same dose of GM-CSF, CpG, and OVA elicited only a moderate Th1 response. Moreover, the MSR vaccine significantly delayed the onset of EG7-OVA tumor growth and extended the survival of the mice compared to the bolus vaccine (GM-CSF + CpG + OVA) (Fig. 6). | ||
| Fig. 6 (A) A schematic representation of the in vivo spontaneous assembly of MSRs and the recruitment of host cells for maturation. (B) Prophylactic cancer vaccine study using injectable MSRs. EG7-OVA tumor volume after the subcutaneous injection of various vaccine formulations ten days before tumor inoculation (n = 10). In (B), the tumor volumes were compared on days 21, 23, and 25 following the onset of tumor growth in the vaccine group. MSR, mesoporous silica microrod; PBS, phosphate buffered saline; OVA, ovalbumin. Adapted from ref. 134 with permission from Springer Nature. | ||
The same group further improved the MSR scaffold-based vaccine system by coating the MSR with cationic polymer polyethyleneimine (PEI).133 The PEI coating promoted the maturation and the antigen cross-presentation level of DCs, which may be due to enhanced lysosome rupture upon PEI uptake. Compared to the MSR vaccine in previous work, the GM-CSF, CpG, and OVA-loaded PEI-coated MSR scaffold (designated as MSR-PEI vaccine) stimulated stronger CD8+ cytotoxic T cell responses. Impressively, with a synthetic long peptide from the E7 oncoprotein of HPV as the antigen, one single intrascapular immunization with the MSR-PEI vaccine resulted in complete regression of tumor growth in 80% of mice with an E7-expressing TC-1 carcinoma. Additionally, all the tumor-free mice were protected from a re-challenging with E7-expressing TC-1 carcinoma, which indicated that a potent immunological memory was generated. Moreover, a pool of B16F10 or MT26 neoantigens could be simultaneously loaded into the MSR-PEI scaffold in one formulation, and this formulated vaccine effectively eradicated lung metastases and synergized with an antibody against cytotoxic T lymphocyte-associated antigen 4 (anti-CTLA4) to delay tumor growth in mice. Thus, this injectable inorganic scaffold may offer a simple and modular platform for the delivery of multiple antigens against different epitopes on a tumor cell surface.
6. Perspective
As illustrated by many examples, injectable macroscale biomaterials have exhibited great promise in promoting local cancer immunotherapy with enhanced efficacy and safety. Applications of such biomaterials have been focused to date on the delivery of various immunomodulators, immune cells, and cancer vaccines. Compared to other commonly used biomaterials (e.g., implantable biomaterials, nano-/micro-biomaterials), this class has some unique advantages, such as facile formulation, surgery-free administration, shape fitness to body cavities, ease of depot size control, sustained local release of cargos, and a reduced toxicity. Studies are currently being performed to better understand the interactions between the immune system and the injectable macroscale biomaterials to further improve the physicochemical properties, such as chemical composition, network density, porosity, stiffness, and degradation products, of those materials for cancer immunotherapy.There are several key challenges to be addressed in the future application of this category of biomaterials for cancer immunotherapy in the clinic. First, intra-/peri-tumoral injection of the biomaterials to deep tumors is challenging, though imaging-guided injection techniques may provide solutions to this issue. Second, on-demand release of immune modulators/cells is highly desired for the exquisite manipulation of immune responses with improved spatiotemporal resolution. The injectable macroscale biomaterials developed to date have mainly depended on the passive release of the cargo via either diffusion- or degradation-dependent mechanisms. Controlled cargo release in response to intrinsic stimuli in the tumor microenvironment, such as low pH and matrix metalloproteinases, or extrinsic stimuli, such as light, magnetic fields, heat, and ultrasound, will be important for intelligent injectable biomaterials. Third, little is known about the in vivo expansion, migration, and functional status of encapsulated immune cells inside injectable macroscale biomaterials, which greatly hampers the application of these biomaterials in immune cell delivery. Finally, good biocompatibility is a prerequisite. Until now, few injectable biomaterials have been tested in the clinic for drug delivery applications (we performed a search in ClinicalTrials.gov). One example is a controlled release formulation of paclitaxel in a PEG-PLGA-based injectable thermosensitive hydrogel (OncoGel™), which has entered Phase II clinical trial for the treatment of recurrent glioma.135 Polysaccharides (hyaluronic acid, alginate, chitosan, etc.), collagen, poly(ethylene oxide)–poly(propylene oxide) (PEO–PPO), and PEG–PLGA block copolymers so far constitute the majority of injectable macroscale biomaterials used in the clinic likely due to their excellent biocompatibility and biodegradability. Collaborations among bioengineers, immunologists, and cancer biologists will potentially address these issues and further advance the development of biomaterial-assisted cancer immunotherapy. We foresee that injectable macroscale biomaterials with sophisticated designs could provide a unique platform for the further development of local cancer immunotherapy, especially for combination therapies through the co-delivery of therapeutics with a synergistic effect.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by Swiss National Science Foundation (Project grant 315230_173243), the Fondation Pierre Mercier pour la science, ISREC Foundation with a donation from the Biltema Foundation, Novartis Foundation for medical-biological Research (17A058), and the École polytechnique fédérale de Lausanne (EPFL).References
- D. N. Khalil, E. L. Smith, R. J. Brentjens and J. D. Wolchok, Nat. Rev. Clin. Oncol., 2016, 13, 273–290 CrossRef CAS PubMed.
- A. Ribas and J. D. Wolchok, Science, 2018, 359, 1350–1355 CrossRef CAS PubMed.
- C. H. June, R. S. O'Connor, O. U. Kawalekar, S. Ghassemi and M. C. Milone, Science, 2018, 359, 1361–1365 CrossRef CAS PubMed.
- C. Boutros, A. Tarhini, E. Routier, O. Lambotte, F. L. Ladurie, F. Carbonnel, H. Izzeddine, A. Marabelle, S. Champiat, A. Berdelou, E. Lanoy, M. Texier, C. Libenciuc, A. M. M. Eggermont, J. C. Soria, C. Mateus and C. Robert, Nat. Rev. Clin. Oncol., 2016, 13, 473–486 CrossRef CAS PubMed.
- A. Marabelle, H. Kohrt, C. Caux and R. Levy, Clin. Cancer Res., 2014, 20, 1747–1756 CrossRef CAS PubMed.
- B. Kwong, S. A. Gai, J. Elkhader, K. D. Wittrup and D. J. Irvine, Cancer Res., 2013, 73, 1547–1558 CrossRef CAS PubMed.
- S. Demaria, C. Vanpouille-Box, S. C. Formenti and S. Adams, OncoImmunology, 2013, 2, e25997 CrossRef PubMed.
- H. Wang and D. J. Mooney, Nat. Mater., 2018, 17, 761–772 CrossRef CAS PubMed.
- N. A. Hotaling, L. Tang, D. J. Irvine and J. E. Babensee, Annu. Rev. Biomed. Eng., 2015, 17, 317–349 CrossRef CAS PubMed.
- J. Weiden, J. Tel and C. G. Figdor, Nat. Rev. Immunol., 2018, 18, 212–219 CrossRef CAS PubMed.
- C. Wang, Y. Ye, Q. Hu, A. Bellotti and Z. Gu, Adv. Mater., 2017, 29, 1–24 Search PubMed.
- S. T. Koshy and D. J. Mooney, Curr. Opin. Biotechnol., 2016, 40, 1–8 CrossRef CAS PubMed.
- Q. Fan, Z. Chen, C. Wang and Z. Liu, Adv. Funct. Mater., 2018, 28, 1–25 Search PubMed.
- A. Hasan, A. Khattab, M. A. Islam, K. A. Hweij, J. Zeitouny, R. Waters, M. Sayegh, M. M. Hossain and A. Paul, Adv. Sci., 2015, 2, 1–18 Search PubMed.
- H. Tan and K. G. Marra, Materials, 2010, 3, 1746–1767 CrossRef CAS.
- B. L. Vernon, Injectable Biomaterials: Science and Applications, Woodhead Pub., Oxford, 2011 Search PubMed.
- P. Smith, A. Kay, L. Dunaway and C. LaShan Simpson, Mater. Technol., 2015, 30, B264–B272 CrossRef CAS.
- Y. Li, J. Rodrigues and H. Tomás, Chem. Soc. Rev., 2012, 41, 2193–2221 RSC.
- L. Yu and J. Ding, Chem. Soc. Rev., 2008, 37, 1473–1481 RSC.
- J. D. Kretlow, L. Klouda and A. G. Mikos, Adv. Drug Delivery Rev., 2007, 59, 263–273 CrossRef CAS PubMed.
- S. R. Van Tomme, G. Storm and W. E. Hennink, Int. J. Pharm., 2008, 355, 1–18 CrossRef CAS PubMed.
- S. B. Stephan, A. M. Taber, I. Jileaeva, E. P. Pegues, C. L. Sentman and M. T. Stephan, Nat. Biotechnol., 2015, 33, 97–101 CrossRef CAS PubMed.
- C. G. Park, C. A. Hartl, D. Schmid, E. M. Carmona, H. J. Kim and M. S. Goldberg, Sci. Transl. Med., 2018, 10, eaar1916 CrossRef PubMed.
- S. A. Chew and S. Danti, Adv. Healthcare Mater., 2017, 6, 1600766 CrossRef PubMed.
- A. Singh and N. A. Peppas, Adv. Mater., 2014, 26, 6530–6541 CrossRef CAS PubMed.
- L. Y. Cheng, X. M. Sun, M. Y. Tang, R. Jin, W. G. Cui and Y. G. Zhang, Plast. Aesthetic Res., 2016, 3, 92–99 CrossRef.
- E. Lih, S. H. Oh, Y. K. Joung, J. H. Lee and D. K. Han, Prog. Polym. Sci., 2015, 44, 28–61 CrossRef CAS.
- D. S. Perrien, C. S. Young, P. P. Alvarez-Urena, D. D. Dean, S. E. Lynch and J. O. Hollinger, Spine J., 2013, 13, 580–586 CrossRef PubMed.
- G. Tchartchian, A. Hackethal, A. Herrmann, B. Bojahr, C. Wallwiener, R. Ohlinger, A. D. Ebert and R. L. De Wilde, Arch. Gynecol. Obstet., 2014, 290, 697–704 CrossRef CAS PubMed.
- R. C. A. Frederickson, Eur. Spine J., 1996, 5, S7–S9 CrossRef PubMed.
- S. Bowman, M. E. Awad, M. W. Hamrick, M. Hunter and S. Fulzele, Clin. Transl. Med., 2018, 7, 6 CrossRef PubMed.
- R. Dong, Y. Pang, Y. Su and X. Zhu, Biomater. Sci., 2015, 3, 937–954 RSC.
- E. A. Appel, J. Del Barrio, X. J. Loh and O. A. Scherman, Chem. Soc. Rev., 2012, 41, 6195–6214 RSC.
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS PubMed.
- M. Ikeda, T. Tanida, T. Yoshii, K. Kurotani, S. Onogi, K. Urayama and I. Hamachi, Nat. Chem., 2014, 6, 511–518 CrossRef CAS PubMed.
- H. Shigemitsu, T. Fujisaku, W. Tanaka, R. Kubota, S. Minami, K. Urayama and I. Hamachi, Nat. Nanotechnol., 2018, 13, 165–172 CrossRef CAS PubMed.
- B. Jeong, S. W. Kim and Y. H. Bae, Adv. Drug Delivery Rev., 2012, 64, 154–162 CrossRef.
- H. J. Moon, D. Y. Ko, M. H. Park, M. K. Joo and B. Jeong, Chem. Soc. Rev., 2012, 41, 4860–4883 RSC.
- M. H. Park, M. K. Joo, B. G. Choi and B. Jeong, Acc. Chem. Res., 2012, 45, 424–433 CrossRef CAS PubMed.
- J. H. Hong, H. J. Lee and B. Jeong, ACS Appl. Mater. Interfaces, 2017, 9, 11568–11576 CrossRef CAS PubMed.
- H. Liu, Y. Cheng, J. Chen, F. Chang, J. Wang, J. Ding and X. Chen, Acta Biomater., 2018, 73, 103–111 CrossRef CAS PubMed.
- Z. Li and J. Guan, Expert Opin. Drug Delivery, 2011, 8, 991–1007 CrossRef CAS PubMed.
- L. Klouda, Eur. J. Pharm. Biopharm., 2015, 97, 338–349 CrossRef CAS PubMed.
- X. Bi and A. Liang, In Situ-Forming Cross-linking Hydrogel Systems: Chemistry and Biomedical Applications, ed. S. B. Majee, ExLi4EvA, 1st edn, 2016, vol. 6, pp. 131–158 Search PubMed.
- C. A. DeForest and K. S. Anseth, Nat. Chem., 2011, 3, 925–931 CrossRef CAS PubMed.
- T. E. Brown and K. S. Anseth, Chem. Soc. Rev., 2017, 46, 6532–6552 RSC.
- S. L. Vega, M. Y. Kwon, K. H. Song, C. Wang, R. L. Mauck, L. Han and J. A. Burdick, Nat. Commun., 2018, 9, 614 CrossRef PubMed.
- B. V. Sridhar, J. L. Brock, J. S. Silver, J. L. Leight, M. A. Randolph and K. S. Anseth, Adv. Healthcare Mater., 2015, 4, 702–713 CrossRef CAS PubMed.
- F. Escobar, K. S. Anseth and K. M. Schultz, Macromolecules, 2017, 50, 7351–7360 CrossRef CAS.
- A. Kumar, R. Mishra, Y. Reinwald and S. Bhat, Mater. Today, 2010, 13, 42–44 CrossRef CAS.
- T. M. A. Henderson, K. Ladewig, D. N. Haylock, K. M. McLean and A. J. O'Connor, J. Mater. Chem. B, 2013, 1, 2682–2695 RSC.
- X. Zhao, B. Guo, H. Wu, Y. Liang and P. X. Ma, Nat. Commun., 2018, 9, 2784 CrossRef.
- S. T. Koshy, D. K. Y. Zhang, J. M. Grolman, A. G. Stafford and D. J. Mooney, Acta Biomater., 2018, 65, 36–43 CrossRef CAS PubMed.
- U. S. Patil, A. V. Jaydeokar and D. D. Bandawane, Int. J. Pharm. Pharm. Sci., 2012, 4, 30–36 CAS.
- L. Milling, Y. Zhang and D. J. Irvine, Adv. Drug Delivery Rev., 2017, 114, 79–101 CrossRef CAS PubMed.
- J. M. Michot, C. Bigenwald, S. Champiat, M. Collins, F. Carbonnel, S. Postel-Vinay, A. Berdelou, A. Varga, R. Bahleda, A. Hollebecque, C. Massard, A. Fuerea, V. Ribrag, A. Gazzah, J. P. Armand, N. Amellal, E. Angevin, N. Noel, C. Boutros, C. Mateus, C. Robert, J. C. Soria, A. Marabelle and O. Lambotte, Eur. J. Cancer, 2016, 54, 139–148 CrossRef CAS PubMed.
- Y. Xie, L. Wei and L. Tang, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2018, 10, 1–23 Search PubMed.
- R. Li, S. Pavuluri, K. Bruggeman, B. M. Long, A. J. Parnell, A. Martel, S. R. Parnell, F. M. Pfeffer, A. J. C. Dennison, K. R. Nicholas, C. J. Barrow, D. R. Nisbet and R. J. Williams, Nanomedicine, 2016, 12, 1397–1407 CrossRef CAS PubMed.
- D. G. Leach, N. Dharmaraj, S. L. Piotrowski, T. L. Lopez-Silva, Y. L. Lei, A. G. Sikora, S. Young and J. D. Hartgerink, Biomaterials, 2018, 163, 67–75 CrossRef CAS PubMed.
- Y. Nishida, S. Ohtsuki, Y. Araie, Y. Umeki, M. Endo, T. Emura, K. Hidaka, H. Sugiyama, Y. Takahashi, Y. Takakura and M. Nishikawa, Nanomedicine, 2016, 12, 123–130 CrossRef CAS PubMed.
- T. Yata, Y. Takahashi, M. Tan, H. Nakatsuji, S. Ohtsuki, T. Murakami, H. Imahori, Y. Umeki, T. Shiomi, Y. Takakura and M. Nishikawa, Biomaterials, 2017, 146, 136–145 CrossRef CAS PubMed.
- M. Nishikawa, Y. Mizuno, K. Mohri, N. Matsuoka, S. Rattanakiat, Y. Takahashi, H. Funabashi, D. Luo and Y. Takakura, Biomaterials, 2011, 32, 488–494 CrossRef CAS PubMed.
- H. Shirota, D. Tross and D. Klinman, Vaccines, 2015, 3, 390–407 CrossRef CAS PubMed.
- C. Bode, G. Zhao, F. Steinhagen, T. Kinjo and D. M. Klinman, Expert Rev. Vaccines, 2011, 10, 499–511 CrossRef CAS PubMed.
- X. J. Loh, V. P. Nam Nguyen, N. Kuo and J. Li, J. Mater. Chem., 2011, 21, 2246–2254 RSC.
- R. Li, Y. Li, Y. Wu, Y. Zhao, H. Chen, Y. Yuan, K. Xu, H. Zhang, Y. Lu, J. Wang, X. Li, X. Jia and J. Xiao, Biomaterials, 2018, 168, 24–37 CrossRef CAS PubMed.
- S. Yu, C. Wang, J. Yu, J. Wang, Y. Lu, Y. Zhang, X. Zhang, Q. Hu, W. Sun, C. He, X. Chen and Z. Gu, Adv. Mater., 2018, 30, 1801527 CrossRef PubMed.
- S. Ishii, J. Kaneko and Y. Nagasaki, Biomaterials, 2016, 84, 210–218 CrossRef CAS PubMed.
- Q. Lv, C. He, F. Quan, S. Yu and X. Chen, Bioact. Mater., 2018, 3, 118–128 CrossRef PubMed.
- A. Fakhari, S. Nugent, J. Elvecrog, J. Vasilakos, M. Corcoran, A. Tilahun, K. Siebenaler, J. Sun, J. A. Subramony and A. Schwarz, J. Pharm. Sci., 2017, 106, 2037–2045 CrossRef CAS PubMed.
- K. Ueda, J. Akiba, S. Ogasawara, K. Todoroki, M. Nakayama, A. Sumi, H. Kusano, S. Sanada, S. Suekane, K. Xu, K. H. Bae, M. Kurisawa, T. Igawa and H. Yano, Acta Biomater., 2016, 29, 103–111 CrossRef CAS PubMed.
- C. Wang, J. Wang, X. Zhang, S. Yu, D. Wen, Q. Hu, Y. Ye, H. Bomba, X. Hu, Z. Liu, G. Dotti and Z. Gu, Sci. Transl. Med., 2018, 10, eaan3682 CrossRef PubMed.
- S. T. Koshy, T. C. Ferrante, S. A. Lewin and D. J. Mooney, Biomaterials, 2014, 35, 2477–2487 CrossRef CAS PubMed.
- J. H. Esensten, J. A. Bluestone and W. A. Lim, Annu. Rev. Pathol.: Mech. Dis., 2017, 12, 305–330 CrossRef CAS PubMed.
- J. P. Veluchamy, N. Kok, H. J. van der Vliet, H. M. W. Verheul, T. D. de Gruijl and J. Spanholtz, Front. Immunol., 2017, 8, 631 CrossRef PubMed.
- K. Newick, S. O'Brien, E. Moon and S. M. Albelda, Annu. Rev. Med., 2017, 68, 139–152 CrossRef CAS PubMed.
- H. R. Mirzaei, A. Rodriguez, J. Shepphird, C. E. Brown and B. Badie, Front. Immunol., 2017, 8, 1850 CrossRef PubMed.
- S. S. Neelapu, S. Tummala, P. Kebriaei, W. Wierda, C. Gutierrez, F. L. Locke, K. V. Komanduri, Y. Lin, N. Jain, N. Daver, J. Westin, A. M. Gulbis, M. E. Loghin, J. F. De Groot, S. Adkins, S. E. Davis, K. Rezvani, P. Hwu and E. J. Shpall, Nat. Rev. Clin. Oncol., 2018, 15, 47–62 CrossRef CAS PubMed.
- C. L. Bonifant, H. J. Jackson, R. J. Brentjens and K. J. Curran, Mol. Ther.–Oncolytics, 2016, 3, 16011 CrossRef CAS PubMed.
- J. N. Brudno and J. N. Kochenderfer, Blood, 2016, 127, 3321–3331 CrossRef CAS PubMed.
- S. S. Neelapu, S. Tummala, P. Kebriaei, W. Wierda, C. Gutierrez, F. L. Locke, K. V. Komanduri, Y. Lin, N. Jain, N. Daver, J. Westin, A. M. Gulbis, M. E. Loghin, J. F. De Groot, S. Adkins, S. E. Davis, K. Rezvani, P. Hwu and E. J. Shpall, Nat. Rev. Clin. Oncol., 2018, 15, 47–62 CrossRef CAS PubMed.
- Y. Hori, P. J. Stern, R. O. Hynes and D. J. Irvine, Biomaterials, 2009, 30, 6757–6767 CrossRef CAS PubMed.
- Y. Hori, A. M. Winans, C. C. Huang, E. M. Horrigan and D. J. Irvine, Biomaterials, 2008, 29, 3671–3682 CrossRef CAS PubMed.
- L. S. Wang, C. Du, W. S. Toh, A. C. A. Wan, S. J. Gao and M. Kurisawa, Biomaterials, 2014, 35, 2207–2217 CrossRef CAS PubMed.
- H. Park, B. Choi, J. Hu and M. Lee, Acta Biomater., 2013, 9, 4779–4786 CrossRef CAS PubMed.
- M. Kurisawa, F. Lee, L. S. Wang and J. E. Chung, J. Mater. Chem., 2010, 20, 5371–5375 RSC.
- A. D. Guerra, O. W. H. Yeung, X. Qi, W. J. Kao and K. Man, Theranostics, 2017, 7, 3732–3744 CrossRef CAS PubMed.
- E. Oh, J. E. Oh, J. W. Hong, Y. H. Chung, Y. Lee, K. D. Park, S. Kim and C. O. Yun, J. Controlled Release, 2017, 259, 115–127 CrossRef CAS PubMed.
- K. K. Goswami, T. Ghosh, S. Ghosh, M. Sarkar, A. Bose and R. Baral, Cell. Immunol., 2017, 316, 1–10 CrossRef CAS PubMed.
- T. Chanmee, P. Ontong, K. Konno and N. Itano, Cancers, 2014, 6, 1670–1690 CrossRef CAS PubMed.
- N. Dehne, J. Mora, D. Namgaladze, A. Weigert and B. Brüne, Curr. Opin. Pharmacol., 2017, 35, 12–19 CrossRef CAS PubMed.
- Y. Xu, Z. Li, X. Li, Z. Fan, Z. Liu, X. Xie and J. Guan, Acta Biomater., 2015, 26, 23–33 CrossRef CAS PubMed.
- N. C. Cheng, W. J. Lin, T. Y. Ling and T. H. Young, Acta Biomater., 2017, 51, 258–267 CrossRef CAS PubMed.
- C. T. Tsao, F. M. Kievit, A. Ravanpay, A. E. Erickson, M. C. Jensen, R. G. Ellenbogen and M. Zhang, Biomacromolecules, 2014, 15, 2656–2662 CrossRef CAS PubMed.
- P. Z. Elias, G. W. Liu, H. Wei, M. C. Jensen, P. J. Horner and S. H. Pun, J. Controlled Release, 2015, 208, 76–84 CrossRef CAS PubMed.
- A. Monette, C. Ceccaldi, E. Assaad, S. Lerouge and R. Lapointe, Biomaterials, 2016, 75, 237–249 CrossRef CAS PubMed.
- R. S. O'Connor, X. Hao, K. Shen, K. Bashour, T. Akimova, W. W. Hancock, L. C. Kam and M. C. Milone, J. Immunol., 2012, 189, 1330–1339 CrossRef PubMed.
- M. Saitakis, S. Dogniaux, C. Goudot, N. Bufi, S. Asnacios, M. Maurin, C. Randriamampita, A. Asnacios and C. Hivroz, eLife, 2017, 6, 1–29 CrossRef PubMed.
- S. H. Van Der Burg, R. Arens, F. Ossendorp, T. Van Hall and C. J. M. Melief, Nat. Rev. Cancer, 2016, 16, 219–233 CrossRef CAS PubMed.
- Z. Tabi and S. Man, Adv. Drug Delivery Rev., 2006, 58, 902–915 CrossRef CAS PubMed.
- T. Fifis, A. Gamvrellis, B. Crimeen-Irwin, G. A. Pietersz, J. Li, P. L. Mottram, I. F. C. McKenzie and M. Plebanski, J. Immunol., 2004, 173, 3148–3154 CrossRef CAS.
- H. Liu, K. D. Moynihan, Y. Zheng, G. L. Szeto, A. V. Li, B. Huang, D. S. Van Egeren, C. Park and D. J. Irvine, Nature, 2014, 507, 519–522 CrossRef CAS PubMed.
- W. A. Li and D. J. Mooney, Curr. Opin. Immunol., 2013, 25, 238–245 CrossRef CAS PubMed.
- J. J. Moon, B. Huang and D. J. Irvine, Adv. Mater., 2012, 24, 3724–3746 CrossRef CAS PubMed.
- D. J. Irvine, M. A. Swartz and G. L. Szeto, Nat. Mater., 2013, 12, 978–990 CrossRef CAS PubMed.
- M. L. Bookstaver, S. J. Tsai, J. S. Bromberg and C. M. Jewell, Trends Immunol., 2018, 39, 135–150 CrossRef CAS PubMed.
- M. Nishikawa, K. Ogawa, Y. Umeki, K. Mohri, Y. Kawasaki, H. Watanabe, N. Takahashi, E. Kusuki, R. Takahashi, Y. Takahashi and Y. Takakura, J. Controlled Release, 2014, 180, 25–32 CrossRef CAS PubMed.
- Y. Umeki, K. Mohri, Y. Kawasaki, H. Watanabe, R. Takahashi, Y. Takahashi, Y. Takakura and M. Nishikawa, Adv. Funct. Mater., 2015, 25, 5758–5767 CrossRef CAS.
- Z. Luo, Q. Wu, C. Yang, H. Wang, T. He, Y. Wang, Z. Wang, H. Chen, X. Li, C. Gong and Z. Yang, Adv. Mater., 2017, 29, 1601776 CrossRef PubMed.
- Y. Ishii-Mizuno, Y. Umeki, Y. Onuki, H. Watanabe, Y. Takahashi, Y. Takakura and M. Nishikawa, Int. J. Pharm., 2017, 516, 392–400 CrossRef CAS PubMed.
- H. Wang, Z. Luo, Y. Wang, T. He, C. Yang, C. Ren, L. Ma, C. Gong, X. Li and Z. Yang, Adv. Funct. Mater., 2016, 26, 1822–1829 CrossRef CAS.
- P. Yang, H. Song, Y. Qin, P. Huang, C. Zhang, D. Kong and W. Wang, Nano Lett., 2018, 18, 4377–4385 CrossRef CAS PubMed.
- Y. Shao, Z. Y. Sun, Y. Wang, B. D. Zhang, D. Liu and Y. M. Li, ACS Appl. Mater. Interfaces, 2018, 10, 9310–9314 CrossRef CAS PubMed.
- Z. Wang, C. Liang, F. Shi, T. He, C. Gong, L. Wang and Z. Yang, Nanoscale, 2017, 9, 14058–14064 RSC.
- T. Wang, D. Wang, H. Yu, B. Feng, F. Zhou, H. Zhang, L. Zhou, S. Jiao and Y. Li, Nat. Commun., 2018, 9, 1–12 CrossRef PubMed.
- Z. Wang, Y. Cai, L. Yi, J. Gao and Z. Yang, Chin. J. Chem., 2017, 35, 1057–1062 CrossRef CAS.
- H. Song, P. Huang, J. Niu, G. Shi, C. Zhang, D. Kong and W. Wang, Biomaterials, 2018, 159, 119–129 CrossRef CAS PubMed.
- G. A. Hudalla, J. A. Modica, Y. F. Tian, J. S. Rudra, A. S. Chong, T. Sun, M. Mrksich and J. H. Collier, Adv. Healthcare Mater., 2013, 2, 1114–1119 CrossRef CAS PubMed.
- J. S. Rudra, Y. F. Tian, J. P. Jung and J. H. Collier, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 622–627 CrossRef CAS PubMed.
- J. Chen, R. R. Pompano, F. W. Santiago, L. Maillat, R. Sciammas, T. Sun, H. Han, D. J. Topham, A. S. Chong and J. H. Collier, Biomaterials, 2013, 34, 8776–8785 CrossRef CAS PubMed.
- R. Xing, S. Li, N. Zhang, G. Shen, H. Möhwald and X. Yan, Biomacromolecules, 2017, 18, 3514–3523 CrossRef CAS PubMed.
- X. Li, A. Galliher-Beckley, H. Huang, X. Sun and J. Shi, Vaccine, 2013, 31, 4508–4515 CrossRef CAS PubMed.
- B. M. Friedrich, D. W. C. Beasley and J. S. Rudra, Vaccine, 2016, 34, 5479–5482 CrossRef CAS PubMed.
- X. Wang, Y. Zhang, W. Xue, H. Wang, X. Qiu and Z. Liu, J. Biomater. Appl., 2017, 31, 923–932 CrossRef CAS PubMed.
- Y. Liu, L. Xiao, K. Il Joo, B. Hu, J. Fang and P. Wang, Biomacromolecules, 2014, 15, 3836–3845 CrossRef CAS PubMed.
- A. J. Highton, A. Girardin, G. M. Bell, S. M. Hook and R. A. Kemp, BMC Immunol., 2016, 17, 1–11 CrossRef PubMed.
- A. J. Highton, T. Kojarunchitt, A. Girardin, S. Hook and R. A. Kemp, Immunol. Cell Biol., 2015, 93, 634–640 CrossRef CAS PubMed.
- Z. Sun, J. Liang, X. Dong, C. Wang, D. Kong and F. Lv, ACS Appl. Mater. Interfaces, 2018, 10, 20315–20325 CrossRef CAS PubMed.
- S. A. Bencherif, R. W. Sands, O. A. Ali, W. A. Li, S. A. Lewin, T. M. Braschler, T. Y. Shih, C. S. Verbeke, D. Bhatta, G. Dranoff and D. J. Mooney, Nat. Commun., 2015, 6, 1–13 Search PubMed.
- T. Y. Shih, S. O. Blacklow, A. W. Li, B. R. Freedman, S. Bencherif, S. T. Koshy, M. C. Darnell and D. J. Mooney, Adv. Healthcare Mater., 2018, 7, 1701469 CrossRef PubMed.
- C. L. L. Chiang, F. Benencia and G. Coukos, Semin. Immunol., 2010, 22, 132–143 CrossRef CAS PubMed.
- J. Copie and A. Dalgleish, Curr. Opin. Mol. Ther., 2010, 12, 14–20 Search PubMed.
- A. W. Li, M. C. Sobral, S. Badrinath, Y. Choi, A. Graveline, A. G. Stafford, J. C. Weaver, M. O. Dellacherie, T. Y. Shih, O. A. Ali, J. Kim, K. W. Wucherpfennig and D. J. Mooney, Nat. Mater., 2018, 17, 528–534 CrossRef CAS PubMed.
- J. Kim, W. A. Li, Y. Choi, S. A. Lewin, C. S. Verbeke, G. Dranoff and D. J. Mooney, Nat. Biotechnol., 2015, 33, 64–72 CrossRef CAS PubMed.
- N. L. Elstad and K. D. Fowers, Adv. Drug Delivery Rev., 2009, 61, 785–794 CrossRef CAS PubMed.
- Y. Li, M. Fang, J. Zhang, J. Wang, Y. Song, J. Shi, W. Li, G. Wu, J. Ren, Z. Wang, W. Zou and L. Wang, OncoImmunology, 2016, 5, 1–12 Search PubMed.
| This journal is © The Royal Society of Chemistry 2019 |