
Enhanced local cancer therapy using a CA4P and CDDP co-loaded polypeptide gel depot†

Abstract
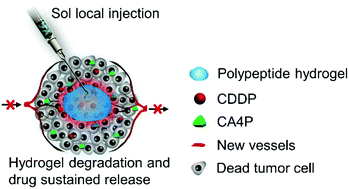
Cancer combination therapy based on drug co-delivery systems provides an effective strategy for enhancing treatment efficacy and reducing side effects. In this work, a new strategy through co-delivery of combretastatin A4 disodium phosphate (CA4P) and cisplatin (CDDP) was developed for the local treatment of colon cancer, through an in situ thermo-gelling hydrogel (mPEG-b-PELG). The results indicated that this material possessed concentration-dependent thermogelling properties and tunable in vivo biodegradability. Also, the drug loaded gel could regulate the in vitro drug release behaviors of both CDDP and CA4P, which promoted the in vivo vessel disrupting effects of CA4P compared with a free drug after local treatment for 48 h. Although the drug co-loaded gel induced less in vitro cell death compared with the free drug co-treated group, this drug co-loaded gel depot showed the highest antitumor efficacy compared with the other experimental groups after peritumoral injection toward C26 tumor bearing mice.
- This article is part of the themed collection: Biomaterials Science Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

