
Peptide ligand-mediated targeted drug delivery of nanomedicines
Changyou
Zhan  *ab
*ab
*ab
Abstract
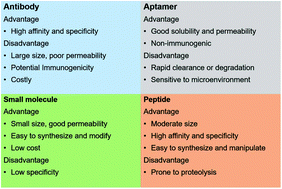
Targeted drug delivery is emerging as a promising strategy to achieve better clinical outcomes. Actively targeted drug delivery that utilizes overexpressed receptors or antigens on diseased tissues is receiving increasing scrutiny, especially due to the uncertainty of existence of the enhanced permeability and retention (EPR) effect in cancer patients. Peptide ligands are advantageous over other classes of targeting ligands due to their accessibility of high-throughput screening, ease of synthesis, high specificity and affinity, etc. In this review, we briefly summarize the resources of peptide ligands and discuss the pitfalls and perspectives of peptide ligand-mediated targeted delivery of nanomedicines.
- This article is part of the themed collection: Biomaterials Science Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

