
Fabrication of redox-responsive doxorubicin and paclitaxel prodrug nanoparticles with microfluidics for selective cancer therapy†

Abstract
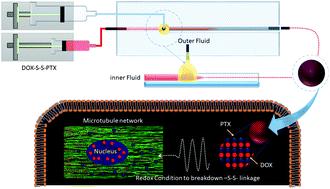
Cancer is an exceptionally confounding disease that demands the development of powerful drug/drugs, without inducing heavy adverse side effects. Thus, different approaches have been applied to improve the targeted delivery of cancer drugs: for example by using nanocarriers. However, nanocarriers are foreign materials, which need further validation for their biocompatibility and biodegradability. In this study, we have chemically conjugated the hydrophilic anticancer drug doxorubicin (DOX) with the hydrophobic drug paclitaxel (PTX) through a redox-sensitive disulfide bond, abbreviated to DOX-S-S-PTX. Subsequently, due to its amphiphilic characterization, the prodrug can self-assemble into nanoparticles under microfluidic nanoprecipitation. These novel prodrug nanoparticles have a super-high drug loading degree of 89%, which is impossible to achieve by any nanocarrier systems, and can be tailored to 180 nm to deliver themselves to the target, and release DOX and PTX under redox conditions, which are often found in cancer cells. By evaluating cell viability in MDA-MB-231, MDA-MB-231/ADR and MEF cell lines, we observed that the prodrug nanoparticles effectively killed the cancer cells, and selectively conquered the MDA-MB-231/ADR. Meanwhile, MEF cells were spared due to their lack of a redox condition. The cell interaction results show that the reduced intermediate of the prodrug can also bind to parent drug biological targets. The hemolysis results show that the nanoparticles are biocompatible in blood. Computer modelling suggested that the prodrug is unlikely to bind to biological targets that parent drugs still strongly interact with. Finally, we confirm that the prodrug nanoparticles have no therapeutic effect in blood or healthy cells, but can selectively eliminate the cancer cells that meet the redox conditions to cleave the disulfide bond and release the drugs DOX and PTX.
- This article is part of the themed collection: Biomaterials Science Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

