
Celastrol-loaded PEG-b-PPS nanocarriers as an anti-inflammatory treatment for atherosclerosis†

Abstract
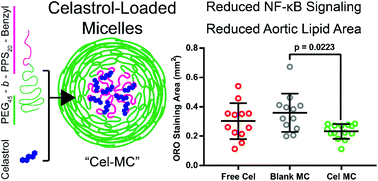
In this work, the hydrophobic small molecule NF-κB inhibitor celastrol was loaded into poly(ethylene glycol)-b-poly(propylene sulfide) (PEG-b-PPS) micelles. PEG-b-PPS micelles demonstrated high loading efficiency, low polydispersity, and no morphological changes upon loading with celastrol. Encapsulation of celastrol within these nanocarriers significantly reduced cytotoxicity compared to free celastrol, while simultaneously expanding the lower concentration range for effective inhibition of NF-κB signaling by nearly 50 000-fold. Furthermore, celastrol-loaded micelles successfully reduced TNF-α secretion after LPS stimulation of RAW 264.7 cells and reduced the number of neutrophils and inflammatory monocytes within atherosclerotic plaques of ldlr−/− mice. This reduction in inflammatory cells was matched by a reduction in plaque area, suggesting that celastrol-loaded nanocarriers may serve as an anti-inflammatory treatment for atherosclerosis.
- This article is part of the themed collection: Biomaterials Science Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

