
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTargeting small molecule drugs to T cells with antibody-directed cell-penetrating gold nanoparticles†
Yu-Sang Sabrina
Yang
ab,
Kelly D.
Moynihan
bc,
Ahmet
Bekdemir
d,
Tanmay M.
Dichwalkar
b,
Michelle M.
Noh
c,
Nicki
Watson
e,
Mariane
Melo
 b,
Jessica
Ingram‡
e,
Heikyung
Suh
b,
Hidde
Ploegh§
e,
Francesco R.
Stellacci
d and
Darrell J.
Irvine
*abcfg
b,
Jessica
Ingram‡
e,
Heikyung
Suh
b,
Hidde
Ploegh§
e,
Francesco R.
Stellacci
d and
Darrell J.
Irvine
*abcfg
aMassachusetts Institute of Technology, Department of Materials Science and Engineering, Cambridge, 02139, USA. E-mail: djirvine@mit.edu
bMassachusetts Institute of Technology, Koch Institute for Integrative Cancer Research, Cambridge, 02139, USA
cMassachusetts Institute of Technology, Department of Biological Engineering, Cambridge, 02139, USA
dÉcole Polytechnique Fédérale de Lausanne, Institute of Materials and Interfaculty Bioengineering Institute, Lausanne, 1015, Switzerland
eWhitehead Institute for Biomedical Research, Cambridge, 02142, USA
fHoward Hughes Medical Institute, Chevy Chase, Maryland 20815, USA
gRagon Institute of MGH, MIT, and Harvard, Charlestown, 02129, USA
First published on 13th November 2018
Abstract
We sought to develop a nanoparticle vehicle that could efficiently deliver small molecule drugs to target lymphocyte populations. The synthesized amphiphilic organic ligand-protected gold nanoparticles (amph-NPs) were capable of sequestering large payloads of small molecule drugs within hydrophobic pockets of their ligand shells. These particles exhibit membrane-penetrating activity in mammalian cells, and thus enhanced uptake of a small molecule TGF-β inhibitor in T cells in cell culture. By conjugating amph-NPs with targeting antibodies or camelid-derived nanobodies, the particles’ cell-penetrating properties could be temporarily suppressed, allowing targeted uptake in specific lymphocyte subpopulations. Degradation of the protein targeting moieties following particle endocytosis allowed the NPs to recover their cell-penetrating activity in situ to enter the cytoplasm of T cells. In vivo, targeted amph-NPs showed 40-fold enhanced uptake in CD8+ T cells relative to untargeted particles, and delivery of TGF-β inhibitor-loaded particles to T cells enhanced their cytokine polyfunctionality in a cancer vaccine model. Thus, this system provides a facile approach to concentrate small molecule compounds in target lymphocyte populations of interest for immunotherapy in cancer and other diseases.
Introduction
Immunotherapies are now an established component of the armamentarium for treating metastatic cancer, with a rapidly growing list of approved drugs and disease indications. Important examples include the checkpoint blockade therapeutics such as antibodies against the negative regulatory receptors PD-1 or CTLA-4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1,2 and chimeric antigen receptor T cell therapy for leukemia.3,4 However, strategies to increase the proportion of patients gaining a benefit from these treatments and/or increasing the durability of immune-mediated tumor regressions are still urgently needed.
1,2 and chimeric antigen receptor T cell therapy for leukemia.3,4 However, strategies to increase the proportion of patients gaining a benefit from these treatments and/or increasing the durability of immune-mediated tumor regressions are still urgently needed.
Much effort to date has focused on targeting cell surface receptors on T cells (and other lymphocytes) with agonistic or blocking antibodies – e.g., checkpoint blockade.5 A complementary approach would be modulation of intracellular signaling pathways directly via small molecule drugs, which might augment T cell function via inhibition of suppressive signaling pathways or by activation of stimulatory signaling nodes. A limitation of such small molecule-based immunomodulation is the likelihood of generalized toxicity as these compounds distribute widely to both tumor antigen-specific and irrelevant lymphocytes throughout the body; such toxicities may be amplified if the signaling pathway of interest in T cells is common to other cell types. Signaling from Transforming Growth Factor-β (TGF-β) provides an example of this challenge: TGF-β is a multifunctional cytokine that regulates cell differentiation and proliferation.6 In the context of tumor immunity, TGF-β is an immunosuppressive cytokine that is often overexpressed in the tumor microenvironment,7,8 and TGF-β signaling inhibits T cell proliferation and effector function.9–15 TGF-β-deficient animals spontaneously reject some tumors,16 and therapeutic inhibition of this pathway has shown efficacy in mouse models of cancer.17–21 However, almost every cell in the body produces TGF-β and has specific receptors for it. The essential role of TGF-β for survival has been illustrated in TGF-β1 knockout mice, 50% of which die in utero, with survivors suffering severe inflammation-related conditions after birth.22,23 Thus, systemic administration of TGF-βi is associated with adverse effects.6,24–26 We previously sought to target a small molecule inhibitor of TGF-β specifically to CD8+ T cells via antibody-targeted liposomes.15 Although these nanocarriers were effective in reversing TGF-β signaling in lymphocytes in vitro, their impact on T cell function in vivo was modest, likely in part due to low drug loading characteristic of these traditional drug delivery vehicles.
Here, we describe a novel lymphocyte-targeted small molecule drug delivery platform, based on very small cell membrane-penetrating amphiphilic gold nanoparticles (amph-NPs). These particles have a 2–3 nm diameter gold core surrounded by an amphiphilic organic ligand shell, a mixed monolayer of alkanethiols terminated by hydrophobic methyl and water-solubilizing sulfonate groups.27 Flexibility of the organic ligands allows these particles to embed within lipid bilayers and transit across bilayers to enter cells in a non-toxic manner.28–31 As described below, we discovered that these particles could also sequester substantial quantities of small molecule drug within the hydrophobic pockets of the ligand shell. This finding led us to hypothesize that conjugation of such drug-loaded particles with whole antibodies or antibody fragments could be used to both (i) temporarily restrict their membrane-penetrating activity and (ii) direct their uptake into specific target cell types. Following binding of the antibody to a target receptor and receptor-mediated endocytosis, proteolysis of the targeting moiety within the endolysosomal pathway would unmask the membrane-penetrating behavior of the particles, allowing subsequent dissemination through the cytoplasm to avoid exocytosis and concentrate the drug throughout the cell. To test this hypothesis, we conjugated both whole antibodies and camelid-derived single-chain antibody fragments to amph-NPs, demonstrated their targeted uptake into CD8+ T cells in vitro and in vivo, and showed the temporal trafficking of these particles to the cytoplasm in lymphocytes. Targeted amph-NPs substantially enhanced uptake of a TGF-β receptor small molecule inhibitor into T cells and promoted enhanced responses to a cancer vaccine, demonstrating a new approach to modulate the function of target cell populations in vivo.
Materials and methods
Nanoparticle synthesis
11-Mercaptoundecane sulfonate ligand (MUS) was synthesized as previously reported.27 All other chemicals were purchased from Sigma-Aldrich and used without further purification. 0.9 mmol gold(III) chloride trihydrate (99.9%) was dissolved in 150 mL of ethanol and 0.75 mmol of ligands (MUS and 1-octanethiol (OT)) at a molar ratio of 1:1 MUS:OT were added to the solution. After 15 minutes of stirring at 900 rpm, an ethanolic solution of sodium borohydride (NaBH4 – 10 times molar excess over gold salt in 150 ml ethanol) was added dropwise to the solution at 25 °C. Black precipitates were observed almost immediately. The final suspension was stirred for an additional 3 hours to ensure reduction of the gold salts. The reaction was quenched by removing the solvent with centrifugation. To remove unreacted chemicals, additional washes with acetone and ethanol were carried out. Finally, water-soluble salts and any residual free ligands were removed using a centrifugal dialysis membrane (Amicon, MWCO 30 kDa). Nanoparticle sizes were characterized by TEM and individual syntheses typically returned mean particle sizes of 2.5–3.5 nm diameter. PEGylated gold nanoparticles (2.1 ± 0.86 nm) coated by thiol-terminated poly(ethylene glycol) (molecular weight 282.35 g mol−1) were purchased from NanoPartz™.
BODIPY fluorescent labeling of gold nanoparticles
Fluorescent dye BODIPY 630/650-X NHS Ester (Invitrogen) and thiol linker (11-mercaptoundecyl amine hydrochloride; Prochimia, Poland) were used as received. 3 mg BODIPY dye and 1.5 mg thiol linker were dissolved in argon-purged amine-free dimethyl formamide and stirred for 6 h in the dark. 3 ml water was added to the solution and stored at 4 °C as a stock solution. To label the nanoparticles with thiol-functionalized Bodipy dye, 10 mg amph-NPs were dissolved in 0.75 ml of water in which 15 μl BODIPY stock solution was added. The reaction was left stirring for 48 h in the dark at 25 °C. Finally, 10 ml acetone was added to the reaction and NPs were washed at least three times to remove unreacted dye via centrifugation for 5 min at 14000 rpm in a tabletop centrifuge.
Transmission electron microscopy imaging of nanoparticles
To analyze size distributions of gold nanoparticles, 10 μL of nanoparticle solution at 2 mg mL−1 was deposited on a carbon coated copper grid, and allowed for 10 min deposition. Residual solution was blotted away using a filter paper and the grid was air-dried. Images were taken under JEOL 2010 FEG Analytical Electron Microscope at 200 kV.Drug loading and quantification
TGF-β inhibitor SB525334 (Selleckchem) was dissolved in ethanol at a concentration of 30 mg mL−1, and mixed with an aqueous suspension of amph-NPs (10 mg mL−1 in water) at a molar ratio of 550 drug molecules per NP. The mixture was placed in a dialysis cassette with 100–500 Da MWCO and stirred against water (3 L dialysis reservoir) for at least 24 hours. To quantify drug loading efficiency, the drug-NP mixture was retrieved from dialysis cassettes 24 later, and free unloaded small molecules were removed using 30k amicon centrifugal tubes at 14kg for 10 minutes. The concentrated suspension of drug-loaded amph-NPs was collected, and a portion of the batch was used for quantification. To extract loaded small molecules from the gold core, 0.05 mg of drug-loaded NP (∼10 μL) solution was mixed with 5 μL of KCN (30 mg mL−1) and 85 μL of ethanol for 4 hours on a shaker to dissolve the gold cores. Clear supernatant was collected and analyzed by a NanoDrop UV-vis spectrometer at a wavelength of 370 nm.Animal care and use
Female C57Bl/6 mice 6–8 weeks of age were purchased from Jackson Laboratories. Animals were cared for in the AAALAC-certified MIT animal facility, and all animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of the NIH and approved by the MIT Institutional Animal Care and Use Committee following local, state, and federal guidelines.Detection of cytosolic TGFβi concentration using HPLC
Primary CD8+ T cells were isolated from spleens of C57Bl/6 mice using an EasySep™ CD8+ T cell isolation kit (Stemcell Technologies). T cells were incubated in complete medium (RPMI containing 10% fetal bovine serum) for 4 h in the presence of amph-NPs loaded with TGF-βi or free TGF-βi. The concentration of drug per cell was then determined by HPLC following cell membrane permeabilization and small molecule extraction in ethanol. To rupture cell membranes, 106 CD8+ T cells were sonicated in a water bath for two minutes at 25 °C. To extract cytosolic TGFβi into the supernatant, 190 μL of ethanol and 10 μL of β-mercapoethanol were added to the cell suspension. The mixture was then placed on a shaker overnight at 25 °C. The resulting cell lysates were centrifuged at 14kg for 10 minutes, and the supernatant containing solubilized TGFbi was loaded into HPLC test vials (80 μL per sample was injected). Samples were run through a reversed phase C18 column (Gemini® 5 μm C18 110 Å, LC Column 250 × 4.6 mm), with a 30 min protocol 20%–95% (Acetonitrile + 0.1% TFA; water + 0.1% TFA). Recovered TGFβi was detected at ∼12.5 min elution time by UV spectroscopy at 350 nm.Kinetics of amph-NP uptake in T cells
Splenocytes from C57Bl/6 mice were isolated and T cells in the cultures were expanded by adding ConA (2 μg mL−1) and IL-7 (1 ng mL−1) in complete RPMI medium for 3 days. Expanded CD8+ T cells were isolated on day 4 by Easysep negative selection and 10 million cells were used per condition. Cells were incubated with 0.1 mg mL−1 of MUSOT amph-NPs at 37 °C for 15 min, 30 min, 4 h, 6 h, and 24 h (triplicate wells per time point) in complete RPMI media in microtiter plates coated with anti-CD3 antibodies and soluble anti-CD28. Free NPs were removed by centrifugation of recovered cells 3 times in PBS, and cell pellets were lysed by the addition of 200 μL aqua regia. Samples were dissolved in aqua regia for two days at 25 °C. Five standards composed of known concentration of MUS/OT amph-NPs were also dissolved in aqua regia in parallel. Prior to analysis, samples were diluted in 2% nitric acid to a total of 4 mL per tube, and then analyzed by inductively coupled plasma-optical emission spectroscopy (ICP-OES, Horiba Activa) to detect total gold content of the samples.Confocal microscopy and flow cytometry
Naïve CD8+ T cells were isolated from the spleens of C57Bl/6 female mice by Easysep negative selection and labeled with carboxyfluorescein succinimide ester (CFSE). Cells were incubated with BODIPY-labeled MUSOT amph-NPs for 3 h at 37 °C. Free NPs were removed by centrifugation and cells were imaged by confocal microscopy (Zeiss LSM 510) using a 63× oil lens. Another portion of cells was analyzed by flow cytometry on a BD FACS Canto.Anti-CD8 VHH nanobody production
The anti-CD8 VHH nanobody (VHH-X118) has been described.32 A Cys-terminated version was designed by removing the C-terminal sortase LPXTG motif from the original construct and introducing a C-terminal cysteine after the polyhistidine tag. The open reading frame was synthesized as a genomic block (Integrated DNA Technologies) and cloned into the pHEN6 bacterial expression vector.33 The resulting plasmid was chemically transformed into WK6 E. coli cells. Following expression, periplasmic extracts were obtained through osmotic shock as previously described,34 and his-tagged VHH protein was purified by affinity chromatography using Ni-NTA agarose beads (ThermoFisher).Antibody or nanobody conjugation
Amph-NPs were mixed with a 120-fold molar excess of 11-Amino-1-undecanethiol hydrochloride (Sigma) at a final NP concentration of 10 mg mL−1 NPs in water and placed on a shaker for 1 hour at 25 °C for ligand exchange. NPs were washed in water two times to remove excess unbound ligands by centrifugation at 14 Kg for 10 minutes using Amicon 30 kDa MWCO filters. A third wash was performed in PBS (pH 7.4). NMR results confirmed that 14% of total ligands on amph-NPs were exchanged by amine ligands. To introduce maleimide groups onto the amino-functionalized NPs, amino-NPs were mixed with a 40-fold molar excess of sulfo-MBS heterobifunctional crosslinkers (Life Technologies) at a final NP concentration of 10 mg mL−1 in PBS (pH 7.4) and placed on a shaker for 1 hour at 25 °C. NPs were washed in PBS (pH 7.4) two times to remove excess unbound linkers by centrifugation at 14 Kg for 10 minutes using Amicon 30 kDa MWCO filters. A third wash was performed in PBS (pH 7.2). The concentration of maleimide-functionalized amph-NPs was measured by UV-vis (Absorbance at 510 nm) using the gold particles’ known extinction coefficient (9.372 L g−1 cm−1) and applying Beer's law. Antibody solution (Anti-Mouse CD8α Clone 53-6.7 or rat IgG2a K Isotype Control purchased from eBioscience) was concentrated to 4 mg mL−1 and mildly reduced by addition of a 25-fold molar excess of dithiothreitol (DTT) in the presence of 10 mM EDTA for 20 minutes at 25 °C. Excess DTT was removed via centrifugation using 7 kDa MWCO desalting columns. Mildly reduced whole antibodies (1 mg mL−1) or VHH-cysteine were immediately coupled with maleimide-functionalized NPs (5 mg mL−1) at a mass ratio of 4:1 NP:protein in PBS (pH 7.2) overnight. Uncoupled free antibodies or VHHs were separated from the particles by centrifugation using an airfuge (Beckman).
Gel electrophoresis analysis of nanoparticle conjugates
The conjugation efficiency of antibody-NP products was quantified by SDS PAGE (NuPAGE™ Novex™ 4–12% bis–tris protein gels purchased from ThermoFisher Scientific). The concentration of airfuge-purified Ab-NPs was determined by UV-vis (Absorbance at 520 nm). Ab-NPs (15–30 μg) were reduced in β-mercapoethanol (βME) for 1–2 days to completely strip ligands from the particles. Supernatant containing 15–30 μg NPs (15 μL) was mixed with 5 μL of 4× SDS PAGE sample loading buffer and heated at 80 °C for 10 minutes. Denatured samples were loaded into gel wells and ran for 40 minutes at 100 kV. Gels were stained with Coomassie blue, imaged using an ImageQuant gel imager and analyzed by ImageJ software.Primary cell isolation and antibody staining for mass cytometry
100 μL of blood was collected via the retro-orbital route, or spleens were collected from C57Bl/6 mice, and splenocytes or peripheral blood mononuclear cells (PBMCs) were isolated. Cell pellets were resuspended in staining buffer followed by antibody staining and fixing: cells were incubated with a selected antibody cocktail (Anti-Mouse CD45 (30-F11)-147Sm; Anti-Mouse CD3e (145-2C11)-152Sm; Anti-Mouse CD8a (53-6.7)-168Er; Anti-Mouse CD4 (RM4-5)-172Yb; Anti-Mouse CD45R/B220(RA36B2)-176Yb; Anti-Mouse CD11b (M1/70)-148Nd; Anti-Mouse Ly-6G (Gr-1) (RB6-8C5)-174Yb; Anti-Mouse CD11c (N418)-142Nd; Anti-Mouse F4/80 (BM8)-159Tb; Anti-Mouse NK1.1 (PK136)-170Er; Anti-Mouse CD64 (X54-5/7.1)-151Eu; Anti-Mouse CD326 [EpCAM] (G8.8)-165Ho) at 25 °C for 30 minutes, excess antibodies were removed by centrifugation, and cells were stained with cell-ID™ Intercalator-Ir in fix and perm solution (detailed protocol available from Fluidigm website. https://www.fluidigm.com/productsupport/cytof-helios). Prior to CyTOF analysis, fixed cells were washed in MaxPar staining buffer twice and MaxPar water once to remove excess iridium. Cells were resuspended at 0.5–1 million per mL in 1:10 calibration beads (EQ™ Four Element Calibration Beads, Fluidigm) in water and 250 μL samples were analyzed by a Fluidigm CyTOF2 at a flow rate of 0.045 mL min−1 or Helios at a flow rate of 0.030 mL min−1.
TEM imaging of CD8+ T cell thin sections
Following amph-NP or antidbody-conjugated NP treatments, cells were washed 2 times with pH 7.4 PBS to remove unbound NPs. The cells were fixed in 2.5% glutaraldehyde + 3% paraformaldehyde with 5% sucrose in 0.1 M sodium cacodylate buffer (pH 7.4) for 1 h at 4 °C, pelleted, and post-fixed in 1% OsO4 in veronal-acetate buffer. The cell pellet was stained in block overnight with 0.5% uranyl acetate in veronal-acetate buffer (pH 6.0), then dehydrated and embedded in Embed-812 resin (Electron Microscopy Sciences). Sections were cut on a Reichert Ultracut E microtome with a Diatome diamond knife at a thickness setting of 50 nm. TEM imaging was conducted with an accelerating voltage of 80 kV using a JEOL 200 CX Transmission Electron Microscope at the Center of Materials Science (CMSE) in MIT.Vaccine studies
Trp1455–463 (TAPDNLGYM), Trp2180–188 (SVYDFFVWL), and gp10020–39 (AVGALEGPRNQDWLGVPRQL) peptide antigens linked to a PEG-DSPE amphiphile were prepared as previously described.35 B16F10 cells (200000) were injected subcutaneously in the flank of C57BL/6J female mice, and trivalent amph-vaccines (10 μg each amph-antigen; Trp1, Trp2, gp100) were administered four days after tumor inoculation s.c. at the tail base along with adjuvant cyclic-di-GMP (Invivogen, 25 μg per mouse). Mice were boosted with the same dose of vaccines 7 days post priming. TGFβi treatment (5 μg per mouse; delivered soluble or loaded in CD8-targeting VHH-amph-NPs) was initiated 8 days post tumor inoculation, and dosed every other day for a total of 8 doses. Intracellular cytokine staining was performed on 20 days post tumor inoculation to measure immune responses against tumor antigens.
Intracellular cytokine staining
Blood from immunized mice was collected (100 μL per mouse) in EDTA-containing tubes, followed by ACK lysis of red blood cells. Cells were pelleted on a 96-well plate and washed with PBS once. Trp1, Trp2, or gp100 peptides (10 μg mL−1) were added to cells in complete RPMI media and incubated for 2 hours in 37 C. Brefeldin A (eBioscience) was added at the recommended dilution (1:1000), and cells were incubated for another 4 hours at 37 °C, for a total of 6 hours with peptides, followed by a single PBS wash. Cells were stained with fixable live/dead aqua (Life Technologies) for 15 minutes at 4 degrees, then washed once with flow cytometry buffer (PBS 1% BSA 5 mM EDTA). Antibodies staining extracellular proteins (CD4, CD8, etc.) were added at 1:100 dilution to the cell pellets and incubated for 15 minutes at 4 °C, followed by a single wash with flow cytometry buffer. Cells were fixed using BD Cytofix for 15 minutes at 4 °C, and washed with BD Perm Wash. Antibodies against IFN-γ and TNF-α were added at a 1:75 dilution in BD Perm Wash for 30 minutes at 4 °C, followed by BD Perm Wash twice. Cells were resuspended in flow cytometry buffer and analyzed on a BD LSR Fortessa.
Results and discussion
Drug loading in amph-NPs and impact of amph-NP-mediated delivery on drug uptake in vitro
Prior studies by the Rotello group have demonstrated that organic layer-capped gold nanoparticles can sequester hydrophobic small molecules in their ligand shell; for example, up to ∼10 small molecules could be loaded into the ligand shell of zwitterionic gold nanoparticles with a 2.5 nm in core diameter.36 Inspired by these findings, we evaluated the loading of a poorly water-soluble TGF-β inhibitor (SB525334, hereafter TGF-βi) into the shell of amph-NPs by co-dissolving drug and particles in 20% ethanol, followed by dialysis to remove the organic solvent and drive the drug into the particle ligand shells (Fig. 1a). UV-vis spectroscopy of soluble TGF-βi solutions showed a distinctive absorption peak at 370 nm (Fig. S1†). Using amph-NPs with a 2.4 ± 0.75 nm core diam. (Fig. S2a and b†), approximately 15 TGF-βi molecules were loaded per NP, as detected by UV-vis spectroscopy (Fig. 1b) or HPLC analysis (not shown). Notably, this drug loading corresponds to an increase in the solubility of the TGF-βi of at least 10-fold. Despite their small size, amph-NPs showed a substantial improvement over our prior studies of liposomes as delivery vehicles for TGF-βi,37 with >10-fold more drug molecules per carrier particle than we achieved with 100 nm-diam. liposomes.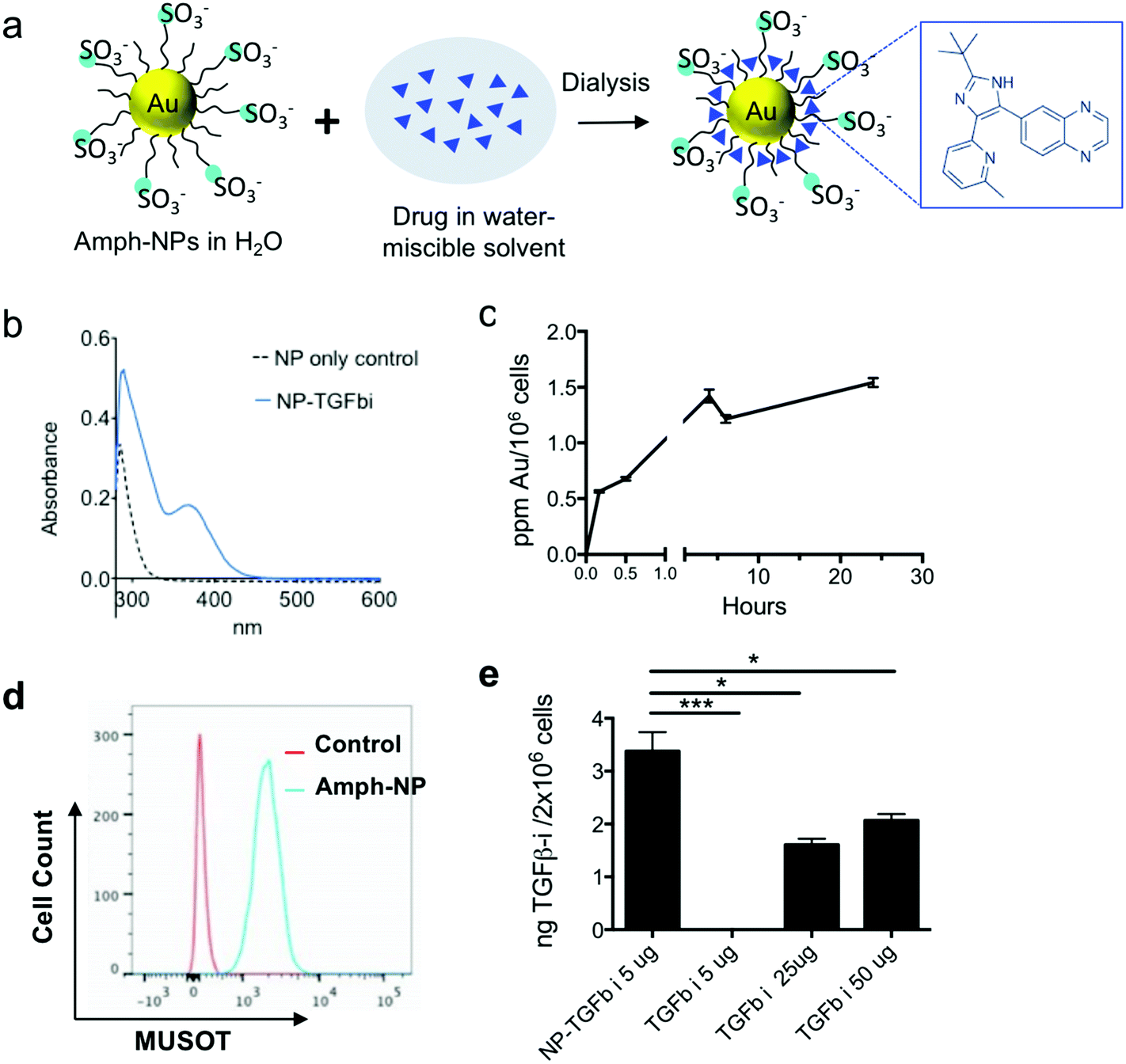 | ||
| Fig. 1 TGF-βi drug loading, quantification and cellular uptake in CD8+ T cells in vitro. (a) Schematic illustration of small molecule TGF-βi sequestration in the amphiphilic ligand shell of amph-NPs. (b) Unloaded amph-NPs or amph-NPs loaded with TGF-βi were treated with β-mercaptoethanol to disrupt the ligand shell followed by UV-vis absorbance measurements to detect released TGF-βi. (c) Primary CD8+ T cells from C57Bl/6 mice were incubated with 0.1 mg mL−1 amph-NPs at 37 °C for varying times and then total gold content was assessed by ICP-OES (n = 3 samples/condition). (d) CD8+ T cells were incubated with 0.1 mg mL−1 BODIPY-labeled amph-NPs at 37 °C for 3 h and then analyzed by flow cytometry, compared to unmanipulated control T cells. (e) Primary murine CD8+ T cells were incubated in serum-containing medium with NP-TGF-βi or free TGF-βi at the indicated drug concentrations for 4 h, followed by HPLC analysis of intracellular TGF-βi concentrations. *, p < 0.05; ***p < 0.001 by ANOVA with Tukey's post test. | ||
Although most hydrophobic small molecule drugs are thought to enter cells by passive diffusion, accumulation of therapeutically functional doses of drugs at intracellular protein targets can still be limiting, even in vitro.38 We hypothesized that small molecules partitioned in the ligand shell of amph-NPs could be transported into cytosolic compartments, as we have previously shown that amph-NPs embed within and penetrate through cell membranes.28–30,39 To test this hypothesis, we first assessed uptake of amph-NPs in primary murine CD8+ T cells. Gold particle accumulation in T cells was detectable within 30 minutes and began to plateau by 3 h as assessed by ICP-OES (Fig. 1c). Parallel analysis of the uptake of fluorophore-conjugated amph-NPs in T cells by flow cytometry showed uniform uptake of particles in the entire T cell population by 3 h (Fig. 1d). To test the impact of amph-NP delivery on uptake of TGF-β inhibitor, CD8+ T cells were incubated with free TGF-βi or amph-NPs loaded with equivalent amounts of the drug (NP-TGF-βi) for 4 hours, and the resulting intracellular TGF-βi concentration was quantified via HPLC analysis of cell lysates. We observed significantly enhanced TGF-βi accumulation in CD8+ T cells in vitro when cell-penetrating amph-NPs were used as a chaperone (Fig. 1e): at 5 μg mL−1 of added free drug, no TGF-βi was detectable in the cells, while the inhibitor was readily detected in T cells treated with NP-TGF-βi. A 10-fold higher concentration of free drug still only reached ∼60% of the levels of intracellular drug accumulated in T cells by the amph-NPs. This result suggested that the membrane penetration properties of amph-NPs can substantially increase the uptake and retention of small molecules intracellularly.
Functionalization of amph-NPs with targeting proteins
Having observed clear benefits to delivery of a small molecule drug into T cells in vitro, we next considered how to direct these particles to specific target cells in vivo. While the membrane-penetrating capacity of amph-NPs confers superior cytosolic delivery capability, keeping them specific to only target cells in vivo is a challenge, because lipid membranes are present on every cell. We hypothesized that by conjugating amph-NPs to antibodies or antibody fragments – which would themselves have sizes comparable to or larger than the nanoparticle itself – we could transiently block the particles’ cell-penetrating behavior, enabling targeted uptake into specific cells. To test this hypothesis, we conjugated intact anti-CD8 antibodies or camelid-derived single-chain VHH targeting moieties to amph-NPs at a molar ratio of approximately 1 protein per particle. Protein conjugation was achieved by first carrying out a ligand exchange reaction of 11-amino-undecanethiol ligands (AUT) with the MUS/OT ligand shell of the particles, and using 1H NMR to quantify changes in the ligand shell composition (Fig. 2a, step 1). The ligand ratio was optimized to achieve a final ligand layer composition of MUS:OT:AUT = 6:3:1, corresponding to 10% ligand replacement (Fig. S3†). Next, the introduced amines were modified with a maleimide functional group using the heterobifunctional linker m-maleimidobenzoyl-N-hydroxysulfosuccinimide ester (Sulfo-MBS, Fig. 2a step 2). Finally, maleimide functionalized amph-NPs were mixed with either dithiothreitol (DTT)-reduced whole anti-CD8 antibodies or anti-CD8 nanobodies (camelid VHHs32) bearing a free cysteine to conjugate targeting proteins to the particles (Fig. 2a step 3). After optimization of reaction conditions, SDS-PAGE analysis showed that ∼1 antibody was conjugated per particle, whereas control particles (without maleimide functionalization) resulted in minimal non-specific binding (Fig. 2b and c). VHH-conjugated particles were similarly prepared with on average 0.77 proteins per particle; non-specific binding of VHH proteins to control amph-NPs without maleimide functional groups was undetectable (Fig. 2d).
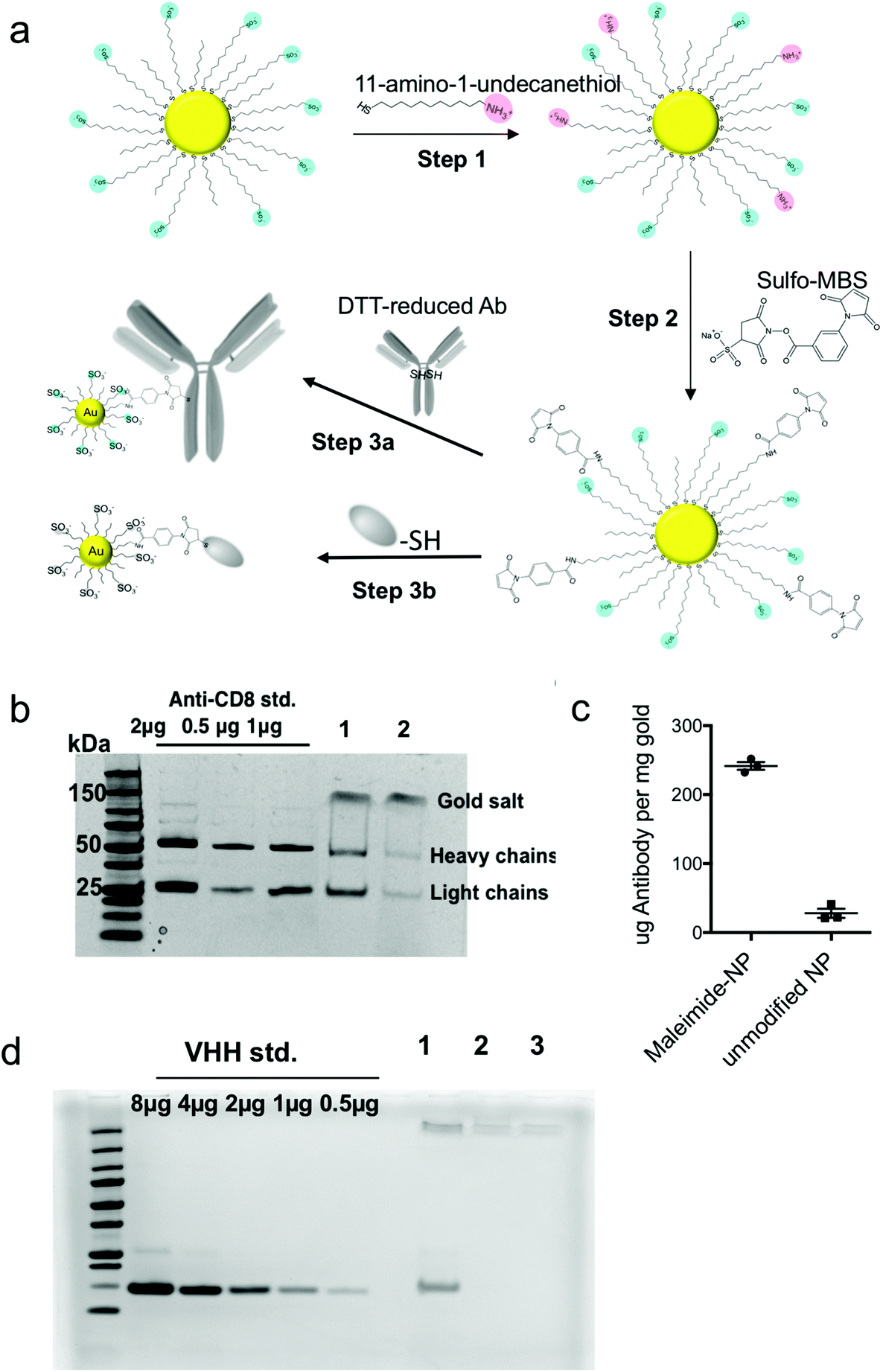 | ||
| Fig. 2 Functionalization of amph-NPs with whole antibodies or nanobodies. (a) Schematic of 3-step process for functionalization of amph-NPs with whole antibodies or VHH nanobodies. (b) SDS-PAGE analysis of anti-CD8 IgG antibody-conjugated amph-NPs (lane 1) and unmodified amph-NPs incubated with antibodies (lane 2). Particles were treated with β-mercaptoethanol to release ligand from the particles prior to PAGE. (c) Quantitative analysis of antibody coupling from SDS-PAGE. (d) SDS-PAGE analysis of anti-CD8 VHH-conjugated amph-NPs (lane 1), unmodified amph-NPs incubated with VHH (lane 2), and unmodified amph-NPs alone (lane 3). Particles were treated with β-mercaptoethanol to release ligand from the particles prior to PAGE. | ||
We recently demonstrated that the biodistribution of metallic nanomaterials such as gold nanoparticles can be precisely quantitated in tandem with deep cellular phenotyping using mass cytometry by time of flight (CyTOF).40 Calibration of the mass cytometer allows the mean number of nanoparticles per cell to be directly calculated. To test whether anti-CD8 antibody or VHH-conjugated amph-NPs would enhance particle accumulation in CD8+ T cells, while reducing off-target uptake in other cell types, we isolated splenocytes from C57BL/6 mice and incubated the cells with anti-CD8-NPs or unmodified control NPs at equivalent gold mass concentrations (10 μg mL−1) for an hour. Unbound NPs were then removed by washing and the cells were stained with metal-conjugated phenotyping antibodies for CyTOF analysis. CD8+ T cells in the splenocyte culture showed a substantial increase in gold uptake per cell and an overall more homogenous gold concentration across the population when incubated with anti-CD8-NPs vs. unfunctionalized nanoparticles (Fig. 3a). By contrast, CD4+ T cells took up both anti-CD8-NPs and unmodified amph-NPs at similar low levels (Fig. 3b). We also observed a decreased intensity of CD8 staining on CD8+ T cells incubated with anti-CD8-NPs, due to competitive binding of the antibody-conjugated nanoparticles with the phenotyping antibody (Fig. 3a). Quantitatively, anti-CD8 functionalization increased amph-NP uptake in CD8+ T cells by 9.2-fold in vitro, and reduced off-target uptake in most other cell types (Fig. 3c). We carried out a similar experiment with anti-CD8 VHH-NPs, incubating unmodified or VHH-conjugated amph-NPs with splenocytes, and characterized uptake in several lymphocyte populations by mass cytometry. As shown in Fig. 3d, VHH conjugation also enhanced uptake of the particles in CD8+ T cells, by 5.3-fold relative to unmodified amph-NPs. (Note that this experiment was performed with a 100-fold lower total particle concentration relative to Fig. 3a–c, hence the lower quantitative amount of particles accumulated in the cells). Thus, uptake into specific target cells was greatly increased by antibody or VHH targeting of amph-NPs in vitro.
 | ||
| Fig. 3 Quantification of antibody-mediated uptake of amph-NPs in lymphocytes in vitro. (a–c) Splenocytes from C57Bl/6 mice were incubated with 10 μg mL−1 (gold mass) amph-NPs or anti-CD8-conjugated amph-NPs for 1 h, then stained with phenotypic antibodies and analyzed by mass cytometry. Shown are representative cytometry graphs overlaying uptake of amph-NPs (blue) and anti-CD8-NPs (red) in CD8+ (a) and CD4+ (b) T cells, and quantitative mean nanoparticle uptake per cell from CyTOF (c, n = 3 samples/condition). (d) Splenocytes were incubated with 0.1 μg mL−1 amph-NPs or VHH-NPs at 37 °C for 1 h then stained and analyzed by mass cytometry. ****, p < 0.0001; ***, p < 0.001; **, p < 0.01; n.s., not significant by ANOVA followed by Tukey's post test. | ||
Intracellular trafficking of antibody-functionalized amph-NPs
To investigate the impact of antibody-mediated targeting on the intracellular trafficking of amph-NPs, we visualized nanoparticle distributions within T cells at serial time points after incubation with the cells by TEM imaging of thin cell sections. Primary murine splenic CD8+ T cells were cultured in the presence of unmodified amph-NPs or anti-CD8-NPs for 30 minutes, after which particles in the supernatants were removed by washing. TEM imaging revealed pronounced localization of anti-CD8-NPs at the surface of T cells following 30 minutes of incubation (Fig. 4a), while free amph-NPs showed little to no surface-bound NPs, although amph-NPs localized to endosomes were observed in some cells (Fig. 4b). We next incubated T cells with anti-CD8-NPs for 30 min, washed the cells to remove unbound particles, and incubated the cells for an additional 4 h or 24 h to allow subsequent cellular internalization of surface-bound particles to occur prior to fixing for TEM imaging. At 4 hours post incubation, most anti-CD8-NPs were entrapped in endosomes, likely via receptor mediated endocytosis (Fig. 4c). Interestingly, we observed cytosolic dispersion of anti-CD8-NPs at 24 h post incubation, implying that amph-NPs can still traverse intracellular membranes despite their initial chemical conjugation to the antibody (Fig. 4d). We expect that once bound and internalized by the target cells, the targeting antibody is proteolyzed in endolysosomes, freeing the amph-NP to subsequently disperse into the cytosol with its drug cargo.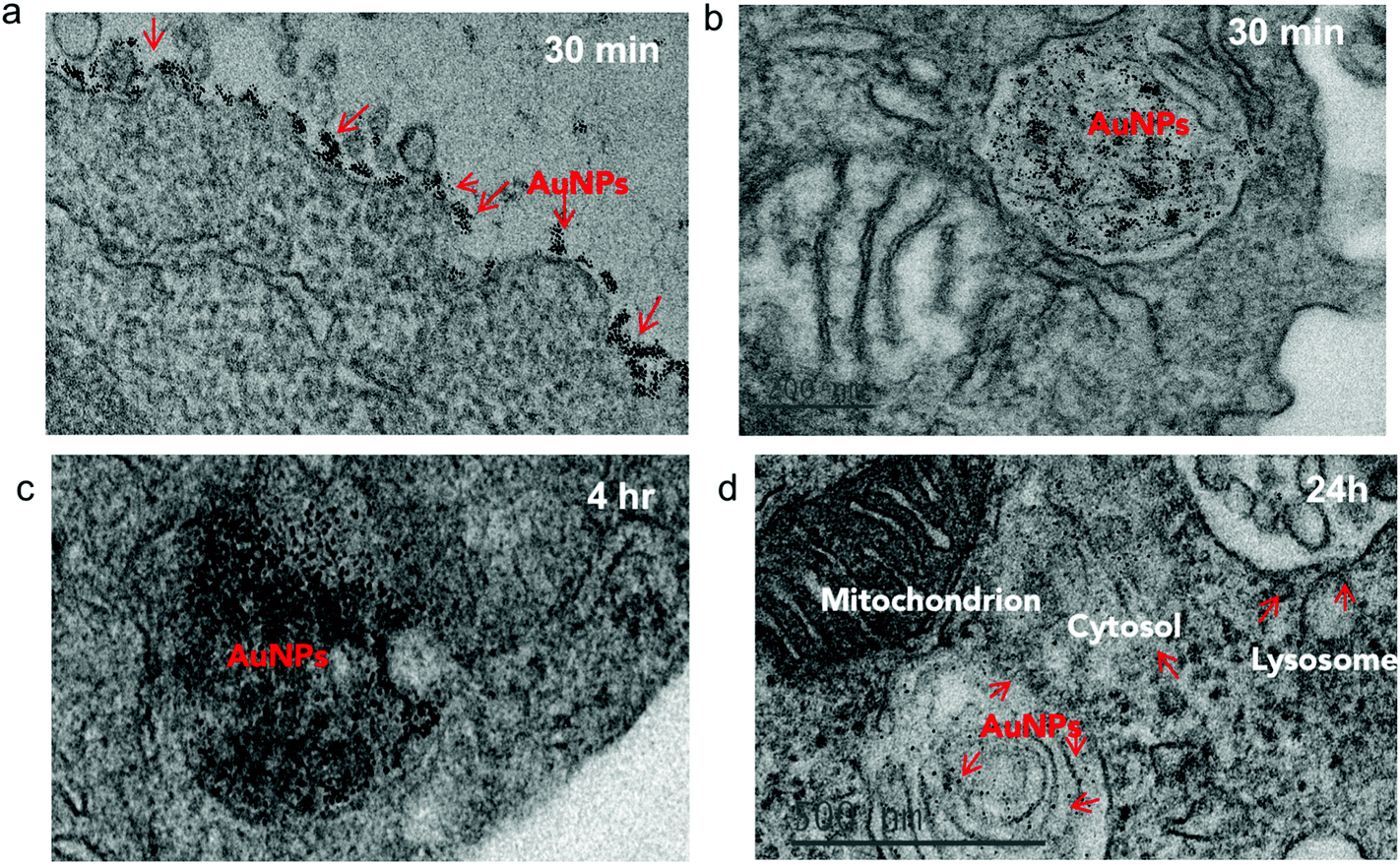 | ||
| Fig. 4 TEM imaging of antibody-targeted amph-NP uptake by CD8+ T cells. T Cells were incubated with 80 μg mL−1 anti-CD8-conjugated amph-NPs (a, c, d) or unmodified amph-NPs (b) for 30 min after which were either fixed immediately (a–b) or incubated in serum-containing media (without NPs) for another 4 hours (c) or 24 hours (d) to allow for NP internalization and dispersion. Shown are representative TEM images of thin sections prepared from each time point. | ||
In vivo targeting of T cells with antibody- and VHH-functionalized amph-NPs
Notably, unlike most published antibody-conjugated nanoparticles, where the particle is significantly larger than the targeting moiety, amph-NPs (∼3–4 nm outer diameter with ligand shell) are smaller than an antibody (∼10 nm). We speculated that this design would result in a more antibody-like biodistribution and pharmacokinetics, promoting nanoparticle targeting efficiency. To test the in vivo targeting efficiency of whole antibody-conjugated particles, anti-CD8-NPs were injected intravenously via the tail vein followed by isolation of peripheral blood mononuclear cells 24 hours post injection for mass cytometry analysis. Anti-CD8 antibody conjugation increased amph-NP uptake in CD8+ T cells over the unmodified particles by 35-fold (Fig. 5a). However, we also observed increased uptake of antibody-conjugated amph-NPs in non-CD8+ cells (ranging from 1.3-fold to 6.7-fold higher than unmodified nanoparticles across different cell types), especially myeloid-derived CD11b+ DCs and CD11b+ F4/80+ macrophages in the blood (Fig. 5a), which we expect may be due to uptake of the whole antibody-conjugated particles via Fc receptors expressed by these cell types.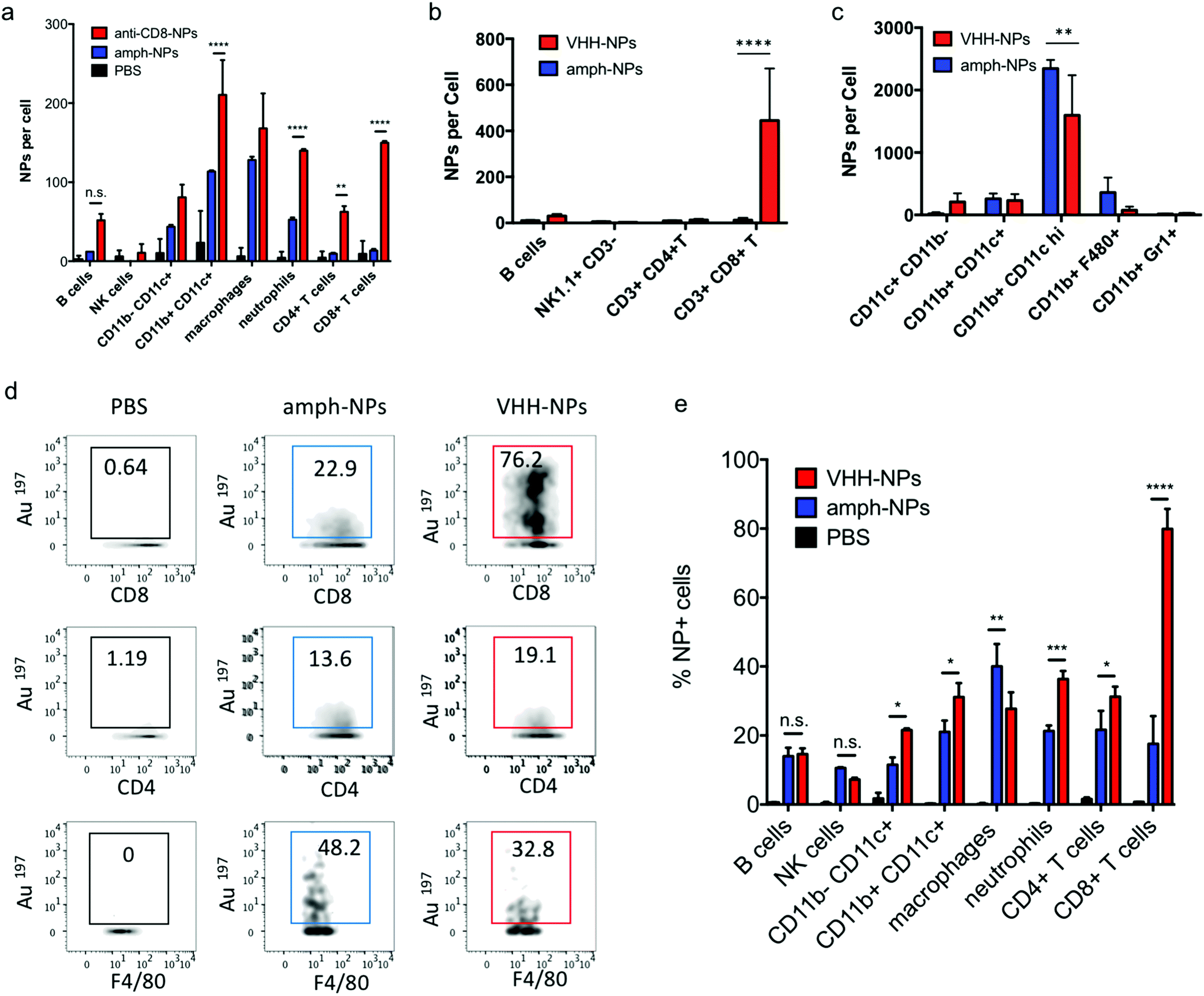 | ||
| Fig. 5 Antibody- and VHH-mediated targeting of nanoparticles to T cells in vivo. (a) C57Bl/6 mice (n = 3 animals/group) were injected i.v. with 150 μg amph-NPs or anti-CD8-NPs, and particle uptake in PBMCs was analyzed 24 h later by mass cytometry. (b–d) C57Bl/6 mice (n = 3 animals/group) were injected i.v. with 100 μg amph-NPs or VHH-NPs. Twenty-four hours later particle uptake in leukocytes was analyzed by mass cytometry. Shown is uptake in peripheral blood lymphocytes (b) and myeloid cells (c), representative mass cytometry plots from several cell populations showing percentages of NP+ cells (d), and quantitation of the mean NP+ cells (e). ****, p < 0.0001; ***, p < 0.001; **, p < 0.01; *, p < 0.05; n.s. not significant by two-way ANOVA with Tukey's multiple comparison test. | ||
To further improve targeting efficiency and reduce uptake in FcR-expressing phagocytes, we next evaluated the use of anti-CD8 VHH at the targeting agent. To evaluate their behavior in vivo, 100 μg unmodified amph-NPs or VHH-amph-NPs were administered intravenously via the tail vein in C57Bl/6 mice. Strikingly, 24 hours post injection, VHH conjugation increased uptake of the nanoparticles in blood CD8+ T cells by 40-fold, from 11 particles per cell to 445 particles per cell, while minimal uptake was observed in other lymphocyte populations (Fig. 5b). Intriguingly, uptake on myeloid cell populations was low for both targeted and untargeted nanoparticles except for a population of CD11b+ CD11chi DCs, where anti-CD8 VHH conjugation resulted in reduced off-target uptake (Fig. 5c). VHH conjugation not only increased the number of NPs per CD8+ cell, but also increased the percentage of CD8+ T cells that contained NPs (Fig. 5d and e). By comparison, VHH-NP showed lower uptake in other cell types such as CD4+ T cells or neutrophils (Fig. 5d and e). Altogether, these data suggest VHH-targeting provided substantially enhanced delivery of amph-NPs to the targeted CD8+ T cell lymphocyte population.
Targeting of TGF-βi to T cells in vivo
Finally, as a preliminary test of the capacity of amph-NPs to deliver drugs to lymphocytes in vivo and functionally impact an immune response, we evaluated the impact of targeting TGF-βi to CD8 cells in the setting of a cancer vaccine. TGF-β signaling inhibits the production of the key effector molecules interferon-γ (IFN-γ), perforin, Granzyme A and B, and Fas ligand by T cells,9–14 and thus we expected that inhibition of this pathway during vaccination could increase T cell effector functions. As a proof of concept, we established a melanoma murine model, and vaccinated the tumor-bearing mice to prime antigen-specific CD8+ T cell responses. Four days after the initial vaccination, mice were repeatedly dosed with soluble TGF-βi (sol. TGF-βi) or TGF-βi loaded in the ligand shell of anti-CD8 VHH-conjugated amph-NPs (VHH-NP(TGF-βi)) (Fig. 6a). Peripheral blood mononuclear cells (PBMCs) were isolated 16 days after the initial vaccination, restimulated by peptide antigens in culture for 6 hours, and analyzed via flow cytometry to detect cytokine-producing antigen-specific T cells. Vaccination alone elicited a clear T cell response to all 3 peptide antigens, with ∼8% of all CD8+ T cells in circulation exhibiting with a polyfunctional (IFN-γ+TNF-α+) response to the Trp1 antigen alone (Fig. 6b and c). Adding treatment with the untargeted free TGF-β inhibitor had no impact on T cell functionality. By contrast, targeting of the drug to T cells with VHH-NPs approximately doubled the proportion of cytokine-producing T cells, increasing production of both IFN-γ and TNF-α by the responding cells (Fig. 6b and c). In addition to increasing the number of cytokine-producing T cells, targeted TGFβi delivery also increased the levels of IFN-γ produced by each cell (Fig. 6d). Unfortunately, the enhanced T cell functionality elicited by targeted TGF-βi therapy in this aggressive tumor model led to only a trend toward slightly slowed tumor growth that was not statistically significant compared to vaccination alone (Fig. S4†); further optimization of treatment regimens for more impactful therapeutic outcomes will be a focus of future work. Vaccination combined with soluble inhibitor treatment trended toward a slightly worse outcome than vaccination alone (Fig. S4†). Thus, targeted TGF-βi delivery clearly enhanced cytokine functionality of T cells over traditional drug therapy in the setting of therapeutic vaccination. | ||
| Fig. 6 TGFβi delivered via VHH-conjugated amph-NPs augments endogenous CD8+ T cell vaccine responses. (a) Timeline of administration of Trp1, Trp2, and gp100 trivalent vaccines and TGFβi administration to tumor-bearing C57Bl/6 mice (n = 5 animals/group). (b) Representative flow cytometry plots of intracellular cytokine staining to detect antigen-specific CD8+ T cell responses from 3 individual mice to each of the 3 peptide antigens in peripheral blood at day 20. (c) Quantitative analysis of percent of IFN-γ- and TNF-α-expressing cells amount total CD8+ T cells. *, p < 0.05; n.s. not significant by ANOVA followed by Tukey's post test. (d) Example histograms (upper panels) and mean fluorescence intensities (MFI, lower panels) of IFN-γ expression in CD8+ T cells after ex vivo antigen restimulation. ***, p < 0.001; **, p < 0.01; *, p < 0.05 by two-tailed unpaired t test. | ||
Conclusions
In conclusion, we demonstrated that small (2–4 nm in diameter) gold nanoparticles with amphiphilic ligand coatings have the dual properties of absorbing hydrophobic small molecules and an ability to disperse in cells via spontaneous lipid membrane penetration or following endocytic uptake by non-disruptively penetrating endosomal membranes. Given these properties, amph-NPs can transport useful doses of drugs across membrane barriers to their cytosolic protein targets to activate or abrogate the corresponding signaling cascades. As an example, we showed that TGF-βi encapsulated in the ligand shell of amph-NPs via hydrophobic–hydrophobic interactions reached a higher intracellular concentration in CD8+ T cells in vitro compared to its free soluble form. Targeted inhibition of TGF-β signaling pathway in circulatory CD8+ T cells in vivo was achieved by antibody or VHH conjugation, which when combined with a vaccine significantly enhanced cytokine production by antigen-specific CD8+ T cells. These data provided a proof of principle for utilizing amph-NPs as an effective lymphocyte-targeted cytosolic small molecule delivery platform. The advantage of this delivery platform is two fold: one, due to the metallic gold core the number of drug carriers per cell can be precisely tracked in vivo for toxicology studies. Two, the capability to solubilize high concentrations of hydrophobic small molecules into amph-NP carrier is superior to conventional lipid or polymer-based approaches. While amph-NPs are designed to target specific lymphocyte populations, a key question is which of these systems is more effective in targeting cells in the circulation vs. tumors vs. lymphoid organs, and answering this will be an important component of future work.Author contributions
Y.S.Y and D.J.I designed the studies and wrote the manuscript. Y.S.Y. conducted the experiments and performed data analyses. Y.S.Y and K.D.M. designed and conducted the vaccine experiments. A.B. and F.S. provided nanoparticles. T.M.D. assisted with tumor measurements. M.M.N. assisted with TEM image analyses, drug loading, and protein nanoparticle conjugations. N.W. assisted with thin-sectioned TEM sample preparations. J.I. and H.P. provided VHH plasmids. M.M. and H.S. assisted designed and produced the cys-terminated VHH.Funding sources
This work was supported in part by the U. S. Army Research Laboratory and the U. S. Army Research Office through the Institute for Soldier Nanotechnologies, under contract number W911NF-13-D-0001, the Melanoma Research Alliance, the Koch Institute Support (core) Grant P30-CA14051 from the National Cancer Institute, and the NIH (awards CA174795 and CA172164). We also acknowledge the EU Horizon2020 FutureNanoNeeds Project.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank Nicole E. Paul for technical assistance with CyTOF sample readout at the Dana-Farber Cancer Institute Boston, MA. We acknowledge the Center for Materials Science and Engineering (CMSE) at MIT for the use of TEM facilities. We thank the Koch Institute Swanson Biotechnology Center for technical support, specifically the Flow Cytometry Core Facility.References
- W. Zou, J. D. Wolchok and L. Chen, Sci. Transl. Med., 2016, 8, 328rv324 CrossRef PubMed
.
- P. Sharma and J. P. Allison, Science, 2015, 348, 56–61 CrossRef CAS PubMed
- C. H. June and M. Sadelain, N. Engl. J. Med., 2018, 379, 64–73 CrossRef CAS PubMed
- M. Sadelain, I. Riviere and S. Riddell, Nature, 2017, 545, 423–431 CrossRef CAS PubMed
- D. M. Pardoll, Nat. Rev. Cancer, 2012, 12, 252–264 CrossRef CAS PubMed
- C. Neuzillet, A. Tijeras-Raballand, R. Cohen, J. Cros, S. Faivre, E. Raymond and A. de Gramont, Pharmacol. Ther., 2015, 147, 22–31 CrossRef CAS PubMed
- R. J. Akhurst and A. Hata, Nat. Rev. Drug Discovery, 2012, 11, 790–811 CrossRef CAS PubMed
- L. Yang, Y. Pang and H. L. Moses, Trends Immunol., 2010, 31, 220–227 CrossRef CAS PubMed
- J. H. Kehrl, L. M. Wakefield, A. B. Roberts, S. Jakowlew, M. Alvarez-Mon, R. Derynck, M. B. Sporn and A. S. Fauci, J. Exp. Med., 1986, 163, 1037–1050 CrossRef CAS PubMed
- S. H. Wrzesinski, Y. Y. Wan and R. A. Flavell, Clin. Cancer Res., 2007, 13, 5262–5270 CrossRef CAS PubMed
- D. A. Thomas and J. Massague, Cancer Cell, 2005, 8, 369–380 CrossRef CAS PubMed
- M. Ahmadzadeh and S. A. Rosenberg, J. Immunol., 2005, 174, 5215–5223 CrossRef CAS
- T. L. Stephen, M. R. Rutkowski, M. J. Allegrezza, A. Perales-Puchalt, A. J. Tesone, N. Svoronos, J. M. Nguyen, F. Sarmin, M. E. Borowsky, J. Tchou and J. R. Conejo-Garcia, Immunity, 2014, 41, 427–439 CrossRef CAS PubMed
- M. K. Donkor, A. Sarkar, P. A. Savage, R. A. Franklin, L. K. Johnson, A. A. Jungbluth, J. P. Allison and M. O. Li, Immunity, 2011, 35, 123–134 CrossRef CAS PubMed
- Y. Zheng, L. Tang, L. Mabardi, S. Kumari and D. J. Irvine, ACS Nano, 2017, 11, 3089–3100 CrossRef CAS PubMed
- L. Gorelik and R. A. Flavell, Nat. Med., 2001, 7, 1118–1122 CrossRef CAS PubMed
- Z. Xu, Y. Wang, L. Zhang and L. Huang, ACS Nano, 2014, 8, 3636–3645 CrossRef CAS PubMed
- J. Liu, S. Liao, B. Diop-Frimpong, W. Chen, S. Goel, K. Naxerova, M. Ancukiewicz, Y. Boucher, R. K. Jain and L. Xu, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 16618–16623 CrossRef CAS PubMed
- J. Park, S. H. Wrzesinski, E. Stern, M. Look, J. Criscione, R. Ragheb, S. M. Jay, S. L. Demento, A. Agawu, P. Licona Limon, A. F. Ferrandino, D. Gonzalez, A. Habermann, R. A. Flavell and T. M. Fahmy, Nat. Mater., 2012, 11, 895–905 CrossRef CAS PubMed
- K. Garrison, T. Hahn, W. C. Lee, L. E. Ling, A. D. Weinberg and E. T. Akporiaye, Cancer Immunol. Immunother., 2012, 61, 511–521 CrossRef CAS PubMed
- S. Takaku, M. Terabe, E. Ambrosino, J. Peng, S. Lonning, J. M. McPherson and J. A. Berzofsky, Int. J. Cancer, 2010, 126, 1666–1674 CAS
- M. M. Shull, I. Ormsby, A. B. Kier, S. Pawlowski, R. J. Diebold, M. Yin, R. Allen, C. Sidman, G. Proetzel and D. Calvin,
et al.
, Nature, 1992, 359, 693–699 CrossRef CAS PubMed
- C. A. Aoki, A. T. Borchers, M. Li, R. A. Flavell, C. L. Bowlus, A. A. Ansari and M. E. Gershwin, Autoimmun. Rev., 2005, 4, 450–459 CrossRef CAS PubMed
- K. Garber, J. Natl. Cancer Inst., 2009, 101, 1664–1667 CrossRef PubMed
- M. J. Anderton, H. R. Mellor, A. Bell, C. Sadler, M. Pass, S. Powell, S. J. Steele, R. R. Roberts and A. Heier, Toxicol. Pathol., 2011, 39, 916–924 CrossRef CAS PubMed
- S. Herbertz, J. S. Sawyer, A. J. Stauber, I. Gueorguieva, K. E. Driscoll, S. T. Estrem, A. L. Cleverly, D. Desaiah, S. C. Guba, K. A. Benhadji, C. A. Slapak and M. M. Lahn, Drug Des., Dev. Ther., 2015, 9, 4479–4499 CAS
- O. Uzun, Y. Hu, A. Verma, S. Chen, A. Centrone and F. Stellacci, Chem. Commun., 2008, 196–198 RSC
- P. U. Atukorale, Y. S. Yang, A. Bekdemir, R. P. Carney, P. J. Silva, N. Watson, F. Stellacci and D. J. Irvine, Nanoscale, 2015, 7, 11420–11432 RSC
- R. C. Van Lehn, M. Ricci, P. H. Silva, P. Andreozzi, J. Reguera, K. Voitchovsky, F. Stellacci and A. Alexander-Katz, Nat. Commun., 2014, 5, 4482 CrossRef CAS PubMed
- R. C. Van Lehn, P. U. Atukorale, R. P. Carney, Y. S. Yang, F. Stellacci, D. J. Irvine and A. Alexander-Katz, Nano Lett., 2013, 13, 4060–4067 CrossRef CAS PubMed
- A. Verma, O. Uzun, Y. Hu, Y. Hu, H.-S. Han, N. Watson, S. Chen, D. J. Irvine and F. Stellacci, Nat. Mater., 2008, 7, 588–595 CrossRef CAS PubMed
- M. Rashidian, J. R. Ingram, M. Dougan, A. Dongre, K. A. Whang, C. LeGall, J. J. Cragnolini, B. Bierie, M. Gostissa, J. Gorman, G. M. Grotenbreg, A. Bhan, R. A. Weinberg and H. L. Ploegh, J. Exp. Med., 2017, 214, 2243–2255 CrossRef CAS PubMed
- K. E. Conrath, M. Lauwereys, M. Galleni, A. Matagne, J. M. Frere, J. Kinne, L. Wyns and S. Muyldermans, Antimicrob. Agents Chemother., 2001, 45, 2807–2812 CrossRef CAS PubMed
- A. Skerra and A. Pluckthun, Science, 1988, 240, 1038–1041 CrossRef CAS PubMed
- K. D. Moynihan, R. L. Holden, N. K. Mehta, C. Wang, M. R. Karver, J. Dinter, S. Liang, W. Abraham, M. B. Melo, A. Q. Zhang, N. Li, S. Le Gall, B. Pentelute and D. J. Irvine, Cancer Immunol. Res., 2018, 6(9), 1025–1038 CrossRef PubMed
- C. K. Kim, P. Ghosh, C. Pagliuca, Z. J. Zhu, S. Menichetti and V. M. Rotello, J. Am. Chem. Soc., 2009, 131, 1360–1361 CrossRef CAS PubMed
- Y. Zheng, M. T. Stephan, S. A. Gai, W. Abraham, A. Shearer and D. J. Irvine, J. Controlled Release, 2013, 172, 426–435 CrossRef CAS PubMed
- P. D. Dobson and D. B. Kell, Nat. Rev. Drug Discovery, 2008, 7, 205–220 CrossRef CAS PubMed
- A. Verma, O. Uzun, Y. Hu, H. S. Han, N. Watson, S. Chen, D. J. Irvine and F. Stellacci, Nat. Mater., 2008, 7, 588–595 CrossRef CAS PubMed
- Y. S. Yang, P. U. Atukorale, K. D. Moynihan, A. Bekdemir, K. Rakhra, L. Tang, F. Stellacci and D. J. Irvine, Nat. Commun., 2017, 8, 14069 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8bm01208c |
| ‡ Deceased. |
| § Current address: Boston Children's Hospital. |
| This journal is © The Royal Society of Chemistry 2019 |