
Spatiotemporal control and modeling of morphogen delivery to induce gradient patterning of stem cell differentiation using fluidic channels
 ab
Daniel A.
Balikov,
cd
Jason X.
Wang,
c
Rizia
Bardhan,
d
Ethan S.
Lippmann
cd
and
Leon M.
Bellan
*abc
ab
Daniel A.
Balikov,
cd
Jason X.
Wang,
c
Rizia
Bardhan,
d
Ethan S.
Lippmann
cd
and
Leon M.
Bellan
*abc
Abstract
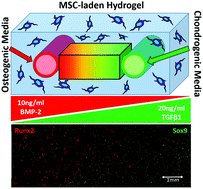
The process of cell differentiation in a developing embryo is influenced by numerous factors, including various biological molecules whose presentation varies dramatically over space and time. These morphogens regulate cell fate based on concentration profiles, thus creating discrete populations of cells and ultimately generating large, complex tissues and organs. Recently, several in vitro platforms have attempted to recapitulate the complex presentation of extrinsic signals found in nature. However, it has been a challenge to design versatile platforms that can dynamically control morphogen gradients over extended periods of time. To address some of these issues, we introduce a platform using channels patterned in hydrogels to deliver multiple morphogens to cells in a 3D scaffold, thus creating a spectrum of cell phenotypes based on the resultant morphogen gradients. The diffusion coefficient of a common small molecule morphogen, retinoic acid (RA), was measured within our hydrogel platform using Raman spectroscopy and its diffusion in our platform's geometry was modeled using finite element analysis. The predictive model of spatial gradients was validated in a cell-free hydrogel, and temporal control of morphogen gradients was then demonstrated using a reporter cell line that expresses green fluorescent protein in the presence of RA. Finally, the utility of this approach for regulating cell phenotype was demonstrated by generating opposing morphogen gradients to create a spectrum of mesenchymal stem cell differentiation states.
- This article is part of the themed collection: Biomaterials Science Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

