
Macromolecular crowding tunes 3D collagen architecture and cell morphogenesis†
 *a
*a
Abstract
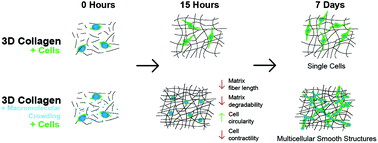
Collagen I is the primary extracellular matrix component of most solid tumors and influences metastatic progression. Collagen matrix engineering techniques are useful for understanding how this complex biomaterial regulates cancer cell behavior and for improving in vitro cancer models. Here, we establish an approach to tune collagen fibril architecture using PEG as an inert molecular crowding agent during gelation and cell embedding. We find that crowding produces matrices with tighter fibril networks that are less susceptible to proteinase mediated degradation, but does not significantly alter matrix stiffness. The resulting matrices have the effect of preventing cell spreading, confining cells, and reducing cell contractility. Matrix degradability and fibril length are identified as strong predictors of cell confinement. Further, the degree of confinement predicts whether breast cancer cells will ultimately undergo individual or collective behaviors. Highly confined breast cancer cells undergo morphogenesis to form either invasive networks reminiscent of aggressive tumors or gland and lobule structures reminiscent of normal breast epithelia. This morphological transition is accompanied by expression of cell–cell adhesion genes, including PECAM1 and ICAM1. Our study suggests that cell confinement, mediated by matrix architecture, is a design feature that tunes the transcriptional and morphogenic state of breast cancer cells.
- This article is part of the themed collection: Biomaterials Science Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

