
Universal nanothin silk coatings via controlled spidroin self-assembly†
 *a
Peyman
Delparastan,
b
Tanner D.
Fink,
a
Joschka
Bauer,
c
Thomas
Scheibel
c
and
Phillip B.
Messersmith
b
*a
Peyman
Delparastan,
b
Tanner D.
Fink,
a
Joschka
Bauer,
c
Thomas
Scheibel
c
and
Phillip B.
Messersmith
b
Abstract
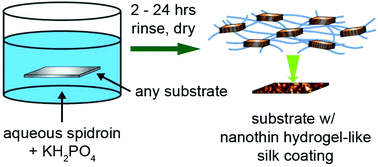
Robust, biocompatible, and facile coatings are promising for improving the in vivo performance of medical implants and devices. Here, we demonstrate the formation of nanothin silk coatings by leveraging the biomimetic self-assembly of eADF4(C16), an amphiphilic recombinant protein based on the Araneus diadematus dragline spidroin ADF4. These coatings result from concurrent adsorption and supramolecular assembly of eADF4(C16) induced by KH2PO4, thereby providing a mild one-pot coating strategy in which the coating rate can be controlled by protein and KH2PO4 concentration. The thickness of the coatings ranges from 2–30 nm depending on the time immersed in the aqueous coating solution. Coatings can be formed on hydrophobic and hydrophilic substrates regardless of surface chemistry and without requiring specialized surface activation. Moreover, coatings appear to be stable through vigorous rinsing and prolonged agitation in water. Grazing incidence wide angle X-ray scattering, single-molecule force spectroscopy, and Congo red staining techniques confirm the formation of β-sheet nanocrystals within the eADF4(C16) coating, which contributes to the cohesive and adhesive stability of the material. Coatings are exceptionally smooth in the dry state and are hydrophilic regardless of substrate hydrophobicity. Under aqueous conditions, nanothin silk coatings exhibit the properties of a hydrogel material.
- This article is part of the themed collection: Biomaterials Science Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

