
Degradable redox-responsive disulfide-based nanogel drug carriers via dithiol oxidation polymerization†

Abstract
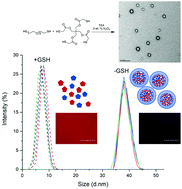
Stimuli-responsive nanogels are important drug and gene carriers that mediate the controlled release of therapeutic molecules. Herein, we report the synthesis of fully degradable disulfide cross-linked nanogel drug carriers formed by oxidative radical polymerization of 2,2′-(ethylenedioxy)diethanethiol (EDDET) as a monomer with different cross-linkers, including pentaerythritol tetramercaptoacetate (PETMA). Because the poly(EDDET) backbone repeat structure and cross-linking junctions are composed entirely of disulfide bonds, these nanogels specifically degrade to small molecule dithiols intracellularly in response to the reducing agent glutathione present inside of cells. Cross-linked nanogels were synthesized using controlled microfluidic mixing in the presence of a nonionic Pluronic surfactant PLU-127 to increase the nanogel stability. Adjusting the monomer to cross-linker ratio from 5 : 1 to 100 : 1 (mol/mol) tuned the cross-linking density, resulting in swelling ratios from 1.65 to >3. Increasing the amount of stabilizing Pluronic surfactant resulted in a decrease of nanogel diameter, as expected due to increased surface area of the resulting nanogels. The monomer to cross-linker ratio in the feed had no effect on the formed nanogel diameter, providing a way to control cross-linking density with constant nanogel size but tunable drug release kinetics. Nanogels exhibited an entrapment efficiency of up to 75% for loading of Rhodamine B dye. In vitro studies showed low cytotoxicity, quick uptake, and fast degradation kinetics. Due to the ease of synthesis, rapid gelation times, and tunable functionality, these non-toxic and fully degradable nanogels offer potential for use in a variety of drug delivery applications.
- This article is part of the themed collection: Biomaterials Science Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

