
Oxygen-rich chemotherapy via modified Abraxane to inhibit the growth and metastasis of triple-negative breast cancer†
Lingtong
Meng‡
a,
Shaoju
Gan‡
a,
Ya
Zhou
a,
Yali
Cheng
a,
Yawen
Ding
a,
Xiaoning
Tong
a,
Jinhui
Wu
 *abc,
Yiqiao
Hu
*abc and
Ahu
Yuan
*abc
*abc,
Yiqiao
Hu
*abc and
Ahu
Yuan
*abc
aState Key Laboratory of Pharmaceutical Biotechnology, Medical School and School of life science, Nanjing University, Nanjing 210093, China. E-mail: wuj@nju.edu.cn; huyiqiao@nju.edu.cn; yuannju@nju.edu.cn
bJiangsu Key Laboratory for Nano Technology, Nanjing University, Nanjing 210093, China
cInstitute of Drug R&D, Medical School of Nanjing University, Nanjing 210093, China
First published on 13th November 2018
Abstract
Abraxane® (Abx), an FDA approved albumin-bound paclitaxel nano-formulation, is one of the most common chemical drugs for the treatment of metastatic triple-negative breast cancer (mTNBC). However, acquired resistance and metastasis are critical factors that limit the treatment of mTNBC by Abx. In particular, both the tumor hypoxic microenvironment and the increase in hydrogen peroxide (H2O2) levels via paclitaxel stimulation primarily mediate the resistance to chemotherapy, where multiple drug resistance proteins such as P-gp and tumor invasion-related cytokines such as VEGF are continuously activated to pump out chemical drugs and aggravate tumor metastasis, respectively. Therefore, it is of great importance to combine tumor oxygenation with commercial chemical drugs for overcoming the acquired resistance and metastasis. In this study, a facile method was developed to deposit manganese dioxide (MnO2) onto the surface of Abraxane® (Abx) to form MnO2-modified Abx (M-Abx). The modification process did not change the critical characteristics of the parent Abx, which might have great potential for application in clinics for the treatment of mTNBC. Tumor oxygenation mediated by M-Abx specifically occurs within the H2O2-overexpressed tumor microenvironment, and significantly downregulates the content of tumor progression-related proteins, such as HIF-1α, P-gp, and VEGF. Ultimately, M-Abx treatment results in about a 2-fold increase in inhibition efficiency of tumor growth in both primary and metastatic tumors compared with traditional Abx therapy. Therefore, oxygen-rich chemotherapy was realized to efficiently sensitize paclitaxel, relieve acquired resistance and inhibit tumor metastasis.
1. Introduction
Metastatic triple-negative breast cancer (mTNBC) is biologically aggressive with poor prognosis.1 Since progesterone receptor, oestrogen receptor and epidermal growth factor receptor-2 are all downregulated on the triple-negative breast tumor cells, endocrine and molecule targeting therapies are not highly effective.2,3 Hence, taxane-based chemotherapy is deemed as the first-line treatment for patients with mTNBC.4,5 Abraxane® (albumin-bound paclitaxel nanoparticle, FDA approved in 2005) is widely used to treat mTNBC.6,7 However, only mild therapeutic outcomes are achieved since multiple issues such as the paclitaxel-mediated acquired resistance limit the treatment of mTNBC with Abraxane® (Abx).8,9It is widely reported that paclitaxel treatment induces tumor cells to generate hydrogen peroxide (H2O2) via NADPH oxidase (NOX) stimulation.10–12 The increase in H2O2 leads to HIF-1α stability, which avoids degradation via the Von Hippel–Lindau (VHL)-related pathway.13,14 HIF-1α further dimerizes with HIF-1β to form HIF-1, which initiates downstream gene transcription.15 As a result, the expression of multidrug resistant proteins, such as P-glycoprotein (P-gp), is greatly increased to pump out paclitaxel.16–18 Simultaneously, vascular endothelial growth factor (VEGF) is significantly upregulated to promote the supply of nutrient to the tumor and metastasis.19,20 Therefore, it is vital to synergize HIF-1α degradation with chemotherapy to achieve optimal therapeutic outcomes for mTNBC treatment.9
The content of HIF-1α is closely related to the level of tumor oxygenation. In the presence of sufficient oxygen, HIF-1α is hydroxylated by prolyl hydroxylases (PHD) to initiate degradation in the proteasome. In addition, H2O2 is another culprit that mediates the stability of HIF-1α, which suppresses the activity of PHD. Inspiringly, manganese dioxide nanoparticles (MnO2-NPs) have been widely studied as a nanoenzyme to convert H2O2 into oxygen molecules strictly within the tumor microenvironment since normal tissue has insufficient H2O2.21–24 Moreover, the released Mn2+ from MnO2-NPs can take advantage of T1-weighted magnetic resonance imaging (MRI) for tumor detection and image-guided therapy.25,26
Albumin is widely used in clinics for treatment of various diseases with guaranteed safety.27,28 In our previous study, we developed a facile method to deposit MnO2 onto albumin to enhance radiotherapy for immobilizing X-ray-induced DNA damage.29 In this study, to integrate tumor oxygenation and chemotherapy into a single system, MnO2 was combined with the commercial nanomedicine Abx to obtain M-Abx in a similar way. The modification does not alter the main characteristics of the parent nanoparticles, including the size, diameter, content and release profile of paclitaxel, which are critical factors in clinical applications. After accumulation within tumor tissues, M-Abx could react with sufficient H2O2 and gradually release oxygen and paclitaxel. The degradation of HIF-1α induced by oxygen improvement and H2O2 consumption leads to the downregulation of P-gp and VEGF, which reduces the efflux of paclitaxel and nutrient supply of the tumor cells, respectively. The suppressed expression of VEGF can also decrease the risk of tumor metastasis. Overall, Abx decorated with MnO2via a one-step facile method can realize oxygen-rich chemotherapy to overcome paclitaxel-mediated acquired resistance and tumor metastasis, which might have great potential for clinical applications (Fig. 1).
 | ||
| Fig. 1 Schematic of the one-step method to prepare M-Abx and the mechanism of inhibiting the growth and metastasis of triple-negative breast cancer via oxygen-rich chemotherapy. | ||
2. Materials and methods
2.1 Materials
Cell-Counting Kit-8 (CCK-8) was purchased from Dojindo Laboratories (Kumamoto, Japan). Coumarin 6 and IR775 were obtained from Sigma-Aldrich (St Louis, MO). Amplex® Red reagent, H2DCFDA, TUNEL assay kit, Ki67 antibody, HIF-1α antibody, VEGF antibody, P-gp antibody, and DAB solution were obtained from Thermo Fisher Scientific (Shanghai, China).2.2 Preparation and characterization of M-Abx
M-Abx was prepared via a facile one-step oxidation method. In brief, 6 mL KMnO4 solution (2.4 mg mL−1) was added to 14 mL Abx (1.2 mg mL−1 of paclitaxel) suspension at 37 °C for 30 min under vigorous stirring. Then, the obtained solution was dialyzed with distilled deionized water three times and condensed with an ultrafiltration device (Millipore 8400, ultrafiltration membrane MW: 30 kDa) to form M-Abx. The final solid product was obtained after freeze drying for 24 h using a freeze dryer (Millrock, RD53, America). Before use, sterile water was added to the product, which was then stored at 4 °C for 30 min to form the M-Abx solution.To track paclitaxel, a mixture comprising coumarin 6 (C6, 0.1 mg mL−1) and paclitaxel (5 mg mL−1) dissolved in an organic solution comprising chloroform and ethyl alcohol was added to 50 mg mL−1 HSA for high pressure homogenization. The organic solution was volatilized in an evaporator to obtain coumarin6@Abx. IR775@Abx was prepared using the same fabrication approach. The diameters and release profiles of the obtained coumarin6@Abx and IR775@Abx were similar with that of Abx (Table S1 and Fig. S1†). Moreover, coumarin 6@M-Abx (C6@M-Abx) and IR775@M-Abx were obtained in a similar way to deposit MnO2 onto the related nanoparticles.
The particle size distribution and zeta potential of M-Abx were measured using Zetasizer Nano (Malvern Instruments, Malvern, UK). The consumption of H2O2 by M-Abx was determined with Amplex® Red reagent according to the manufacturer's instructions. Briefly, after H2O2 (100 μM) reacted with M-Abx ([MnO2] = 0, 20, 40, 60, 80, 100, and 120 μM), the test mixture comprising Amplex® Red reagent and horse radish peroxidase (HRP) was added. Fluorescence spectra of Amplex® Red (Ex = 530 nm) were detected using a microplate reader (Molecular Devices M3, America). The oxygen generating ability of M-Abx was determined using a Clark Oxygen Probe (OX25, Unisense, Denmark). The oxygen probe was immersed in the M-Abx ([MnO2] = 400 μM) or equivalent Abx solution for several minutes to balance the test system. Then, 100 μM H2O2 was injected into the test solutions. After continuous monitoring for 10 min, H2O2 was injected again, and this cycle was repeated four times.
The release of paclitaxel or IR775 was determined in deionized water containing 1.8% (m/v%) β-cyclodextrin. The concentration of paclitaxel was analyzed via high performance liquid chromatography (HPLC, LC20-AT, SHIMADZU, Japan) equipped with a C18 column (Ab = 228 nm). The mobile phase comprised 50% acetonitrile and 50% distilled deionized water. Flow rate was controlled at 1 mL min−1. To inspect the paclitaxel content in the Abx and M-Abx solutions, 950 μL of extraction solution (acetonitrile) was added to 50 μL of sample. After vigorous shaking and ultrasonic incubation, the mixtures were centrifuged at 5000 rpm for 5 min. Then, 10 μL of supernatant was injected into the HPLC system for detection. To avoid effects from the column, an equal amount of H2O2 was added to consume MnO2. The content of IR775 was analyzed using a microplate reader (Ab = 775 nm).
2.3 Generation and consumption of hydrogen peroxide in vitro
4T1 cells were obtained from Procell Life Science & Technology Co., Ltd (Wuhan, China). In order to investigate the H2O2 generation ability of Abx and H2O2 consumption by M-Abx, 4T1 cells with a density of 7 × 104 per well were seeded in 24-well plates covered with glass disks. After attachment, the cells were incubated with Abx and an equivalent amount of M-Abx ([paclitaxel] = 5 μM) for 12 h. The cells were washed three times with PBS. Then, H2DCFDA was applied to indicate the H2O2 contents according to the manufacturer's instructions, and images were captured by an Olympus FV3000 laser scanning confocal microscope.The increase in H2O2 generation of Abx, which was dependent on the incubation time, and the potential of M-Abx for H2O2 consumption were further confirmed by the flow cytometry assay. In brief, 4T1 cells were seeded with a density of 2 × 105 per well in 6-well plates. After incubation for different times (4, 12, and 24 h), Abx and equivalent M-Abx ([paclitaxel] = 5 μM) were removed and stained with the H2O2 detection probe, and then analyzed via flow cytometry (FL1 channel). Moreover, various concentrations of Abx or equivalent M-Abx ([paclitaxel] = 0, 5, 10, 25, and 50 μM) were applied to inspect whether H2O2 generation was dependent on paclitaxel concentration. Equivalent HSA (vehicle) was set as the control.
2.4 Cytotoxicity and penetration of M-Abx in vitro
In order to investigate the enhancement in the therapeutic effect of M-Abx, 4T1 cells with a density of 5 × 103 per well were seeded in 96-well plates. After attachment, the vehicle, MnO2-HSA, Abx and equivalent M-Abx ([paclitaxel] = 0.5 μM, [MnO2] = 2.5 μM) were treated for 36 h, respectively. After the incubated drugs were removed, the treated cells were cultured for additional 72 h. Then, the CCK-8 test solution was added to each well and incubated for 2 h. Finally, the absorbance at 450 nm was used to calculate the relative cytotoxicity, where cells treated with the vehicle (HSA) were set as the control. To observe the enhanced tumor therapy of M-Abx visually in vitro, the cell clone assay was used. After different treatments in the cytotoxicity experiment, 2 × 103 4T1 cells were seeded into 6-well plates. As macroscopic cell colonies formed, the crystal violet staining solution was used to display the therapeutic efficiency.For the detection of the penetration of M-Abx in 3D cell spheroids, 2 × 103 4T1 cells were seeded into ultra-low attachment round bottom 96-well plates (Corning, 7007, America). After cell spheroid formation, free coumarin 6 (C6) or coumarin 6@M-Abx (C6@M-Abx) was added for 8 h incubation. Then, the drugs were washed away using PBS. Fluorescence images were obtained using an Olympus FV3000 laser scanning confocal microscope and analyzed with Nikon NIS-Elements.
In order to inspect the therapeutic effect of M-Abx in vitro, 3D cell spheroids were formed with a density of 3 × 104 per well. Different treatments including vehicle (HSA), MnO2-HSA, Abx and equivalent M-Abx ([paclitaxel] = 0.5 μM, [MnO2] = 2.5 μM) were administered on day 0 and day 3, respectively. The images and diameters of the cell spheroids were recorded and analyzed using Nikon NIS-Elements every other day. The volume of the spheroids was calculated according to following formula: 4/3 × π × (d/2)2.
2.5 Tumor accumulation and overcoming tumor resistance in vivo
Female Balb/c mice were purchased from Yangzhou University Medical Center (Yangzhou, China) and received care in accordance with the Institution Animal Care and Use Committee (IACUC) of Nanjing University. All in vivo experiments were approved and conducted under the guidelines of IACUC of Nanjing University. To investigate the accumulation of M-Abx in the 4T1 breast cancer model, the 5 × 105 4T1 cell suspension was subcutaneously implanted in the flanks of the mice. Sixteen days later, the tumor mass was isolated and cut into small pieces of about 1 mm3, which were subcutaneously implanted in the right axilla of the mice to establish the 4T1 breast cancer model. When the tumor volume reached about 150 mm3, IR775@M-Abx was intravenously injected into the tumor bearing mice. An IVIS Lumina imaging system (Xenogen Corporation-Caliper, Alameda, CA, USA, Ex = 745 nm, Em = 820 nm) was applied to detect the fluorescence of IR775 in the 4T1 tumor bearing mice. After anesthetization, images were obtained from 0.5 h to 72 h after intravenous injection of IR775@M-Abx. For the tissue distribution assay, the mice were sacrificed at 10 h post-injection, and their major organs were isolated and imaged in the same way. Data was analyzed with the IVIS Living Imaging Software.To inspect the H2O2 consumption by M-Abx, the mice bearing 4T1 tumor (∼150 mm3) were sacrificed 4 h after intratumoral injection of 50 μL vehicle, MnO2-HSA, Abx and equivalent M-Abx. Then, the tumor tissues were isolated and weighed. After the addition of tissue lysate, the tumor masses were homogenized with an IKA-Ultra Turrax T25 homogenizer. After centrifugation at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 5 min, the concentration of H2O2 in the supernatant were investigated using the Hydrogen Peroxide Assay Kit (Nanjing Jiancheng Engineering Institute) according to the manufacturer's instructions. All procedures were performed at 4 °C in the dark.
000g for 5 min, the concentration of H2O2 in the supernatant were investigated using the Hydrogen Peroxide Assay Kit (Nanjing Jiancheng Engineering Institute) according to the manufacturer's instructions. All procedures were performed at 4 °C in the dark.
2.6 Immunofluorescence and immunochemistry
To inspect the tumor oxygenation by M-Abx, HIF-1α antibody was applied to stain the hypoxic marker HIF-1α. Mice bearing 4T1 tumor were sacrificed at 36 h after intravenous injection of the indicated solutions. The tumor masses were collected and embedded into tissue freezing medium, and then sliced into a thickness of 6 μm. The slices were stained with HIF-1α antibody (Dylight 488) diluted at 1:200 with 3% BSA at 4 °C for 12 h. In addition, the tumor tissues collected at 48 h post different treatments were used to investigate the expressions of VEGF and P-gp. In brief, the corresponding primary antibodies were incubated with tumor slices for 12 h at 4 °C. Secondary antibodies conjugated with Alexa Fluor® 594 were then added to react for 40 min at 37 °C.
To evaluate the enhanced effect of chemotherapy, Ki67 antibody and the TUNEL assay kit were applied to stain the proliferative and apoptotic cells, respectively. Tumor slices were prepared from mice sacrificed at 36 h after chemotherapy treatments. Immunofluorescence for TUNEL assay and immunochemistry for Ki67 characterization were achieved according to the manufacturer's instructions. All the images of tumor sections were obtained using a Nikon Eclipse Ti and analyzed with Nikon NIS-Elements.
2.7 Enhanced chemotherapy in 4T1 primary and secondary tumor
4T1 tumor bearing mice (∼25 mm3) were divided into four groups, including HSA (vehicle), MnO2-HSA, Abx, and M-Abx ([paclitaxel] = 20 mg kg−1, [MnO2] = 10 mg kg−1). Treatments were performed on day 0, 3, and 7, respectively. Tumor size and body weight were recorded every day, and tumor volume was calculated according to the formula: width2 × length/2. On day 15, the primary tumor masses were removed through surgery. The skin tissues surrounding the tumor lesion were also removed to minimize the risk of primary tumor relapse. On day 44, lung and liver tissues were collected after sacrifice. After fixation in Bouin's solution, the metastatic lesions were observed and further analyzed with hematoxylin and eosin (H&E) staining.In another separate experiment, blood for serum biochemistry was collected after the same protocol of chemotherapy to inspect the influence of the different treatments on 4T1 tumor bearing mice. Serum biochemistry data was measured using a Chemray 430 (Rayto, China). The liver function markers included alanine aminotransferase (ALT), and aspartate aminotransferase (AST), while blood urea nitrogen (BUN) was used to evaluate kidney function. Direct bilirubin (DB), total bilirubin (TB) and the ratio of DB/TB were determined to investigate hemolysis. Furthermore, H&E staining of the major organs was performed on day 15.
In order to inspect the metastasis inhibition of M-Abx directly, an experimental metastatic model was also established. In brief, 5 × 105 4T1 cells suspended in PBS were intravenously injected into the mice. Chemotherapy was performed on day 13 and day 21, and the average survival time and survival rate were recorded.
2.8 Statistical analysis
Statistical significance was calculated using the two-tail Student's t-test for two groups and one-way analysis of variance for multiple groups. A value of p < 0.05 was considered statistically significant.3. Results
3.1 Characterization of M-Abx
To achieve oxygen-rich chemotherapy, a one-step oxidation method was established to prepare MnO2-deposited Abraxane® (termed as M-Abx). Due to the modification of MnO2, the blue-white nanoparticle solution turned brown. After freeze drying, the solid state of M-Abx exhibited long-term stability. In addition, after restoration with sterile water, the main characteristics of M-Abx remained constant compared with the state of the solution before freeze drying (Fig. 2a). | ||
| Fig. 2 Characteristics of M-Abx. (a) Photographs of Abx, M-Abx and their respective freeze dried products. (b) Dynamic light scattering data and polydispersity index. (c) High performance liquid chromatography (HPLC) data of Abx and M-Abx. The insert presents the retention time (RT) and area under curve (AUC) for paclitaxel. (d) Release profile of paclitaxel in vitro (n = 3). (e) Fluorescence changes in the mixture of Amplex® Red and HRP induced by H2O2 (100 μM) reacting with different concentrations of M-Abx ([MnO2] = 0, 20, 40, 60, 80, 100, and 120 μM). (f) Accumulated oxygen generation over time. The arrows indicate the addition of H2O2 solution. Data is shown as mean ± SD. | ||
The mean size of M-Abx slightly increased from 130 nm to 140 nm, which indicated that the facile deposition of MnO2 exhibited negligible influence on the size distribution of the nanoparticles (Fig. 2b). Then, HPLC was applied to detect the impact of MnO2 deposition on paclitaxel. The unchanged retention time of paclitaxel indicated that the deposition of MnO2 did not have any influence on the structure of the loading drug. The similar area under the curves also suggested that the modification of Abx did not affect the content of paclitaxel (Fig. 2c). In addition, the release of paclitaxel from M-Abx was parallel with that of Abx, which might retain similar pharmacokinetic behavior (Fig. 2d). Furthermore, the presence of MnO2 in M-Abx was confirmed via inductively coupled plasma emission spectroscopy (ICP), X-ray photoelectron spectroscopy (XPS) and transmission electron microscopy (TEM) (Table S2, Fig. S2 and S3†). All the above mentioned results indicate that the developed one-step method to deposit MnO2 on the surface of Abx does not have a significant influence on the critical characteristics of parent Abx, which has great potential application for the treatment of mTNBC.
In order to investigate the potency of M-Abx for utilizing H2O2 to generate O2, Amplex® Red reagent was first applied to detect the H2O2 consumption by M-Abx. As the concentration of M-Abx (MnO2) increased, the fluorescence spectrum of Amplex® Red reagent gradually decreased, indicating the ability of M-Abx to consume H2O2 (Fig. 2e). These results suggest that the simple deposition of MnO2 endowed Abx with the ability to generate oxygen in the tumor region because a significantly increased oxygen generation occurred cumulatively after H2O2 addition (Fig. 2f).
3.2 Generation and consumption of hydrogen peroxide in vitro
As shown in Fig. 3a, the green fluorescence of the H2O2 detection probe significantly increased after Abx treatment compared with vehicle treatment. Moreover, the generation of H2O2 was dependent on both the concentration and incubation time of paclitaxel. After the introduction of MnO2 to Abx, the production of H2O2 was significantly suppressed (Fig. 3b and c). | ||
| Fig. 3 Generation and consumption of hydrogen peroxide in vitro. (a) Images of 4T1 cells stained with H2DCFDA and DAPI after the indicated treatments, scale bar = 50 μm. (b) H2O2 content in 4T1 cells with different treatments for the indicated time, as analyzed via flow cytometry. (c) Dynamic changes in H2O2 content as a function of paclitaxel concentration. (d) Cytotoxicity of vehicle, MnO2-HSA, Abx and M-Abx, respectively. (e–f) Photographs and quantification of cell clones in the indicated groups. Data is shown as mean ± SD (n = 3). *p < 0.05; **p < 0.01; and ***p < 0.001. | ||
Next, we investigated the cell viabilities of the different treatments. MnO2-HSA did not impact the survival and proliferation of 4T1 cells. Meanwhile, the cell viability in the M-Abx group decreased to 62.22% compared with that from Abx treatment (79.91%), which might be attributed to the improved paclitaxel resident in the tumor cells (Fig. 3d). Furthermore, the clone assay confirmed the superiority of the improved chemotherapy from M-Abx. As shown in Fig. 3e, abundant macroscopic cell clones were formed in the vehicle and MnO2-HSA groups. Fewer scattered cell clones were observed after M-Abx treatment compared with Abx treatment. The average clone count of 13 clones in the M-Abx group was significantly less than that in the Abx group (49 clones), which indicated the potential of M-Abx nanoparticles for mTNBC chemotherapy (Fig. 3f).
3.3 Penetration and therapeutic effect of M-Abx in 3D cell spheroids
In order to investigate the penetration of M-Abx in tumor tissues, three dimensional (3D) 4T1 cell spheroids were established. To indicate the location of M-Abx, coumarin 6@M-Abx (C6@M-Abx) was applied. As shown in Fig. 4a, the 4T1 cell spheroids remained dim from the 0 μm section to 50 μm section after the free coumarin 6 (C6) treatment, where the green fluorescence only emerged at the margin regions. However, after incubation with coumarin 6@M-Abx, both the marginal area and core region were significantly lit by the fluorescence of coumarin 6 (Fig. 4a). The dynamic intensity profile of coumarin 6 at the 30 μm section also indicates the favorable potential of M-Abx to deliver loaded drugs into the depth of tumor tissue (Fig. 4b). Then, we inspected the therapeutic effect of M-Abx in 3D cell spheroids. Unexpectedly, Abx showed only slight tumor suppression (10.8%) compared with the vehicle group. In contrast, the volume of the cell spheroids significantly decreased after the M-Abx treatment, where 65.9% inhibition of tumor spheroid growth was achieved (Fig. 4c and d).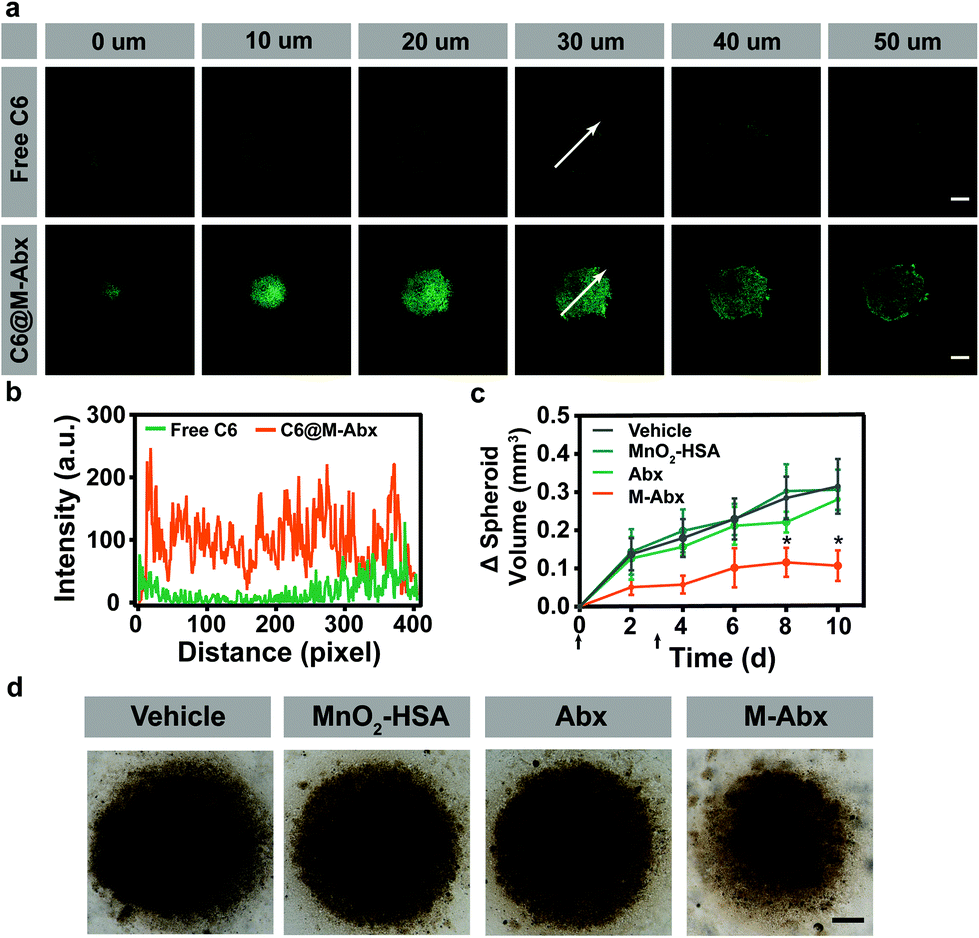 | ||
| Fig. 4 Enhancement of the therapeutic effect of M-Abx in 3D cell spheroids. (a) Penetration of M-Abx in 4T1 cell 3D spheroids, scale bar = 100 μm. (b) Dynamic coumarin 6 fluorescence intensity profiles with the 30 μm sections labeled with white arrows. (c) Dynamic volume of 4T1 cell spheroids. The indicated treatments were performed on day 0 and 4 (n = 3). (d) Photos of 4T1 cell spheroids after the different treatments on day 10, scale bar = 250 μm. Data is shown as mean ± SD. *p < 0.05; **p < 0.01; and ***p < 0.001. | ||
3.4 Tumor accumulation and overcoming tumor resistance in vivo
To determine the accumulation of M-Abx in vivo, a mice bearing 4T1 tumor model was established, as shown in Fig. 5a. When the tumor volume reached about 150 mm3, intravenous injection of IR775@M-Abx was performed. The NIR fluorescence of IR775 progressively increased in the tumor region and peaked at 10 h post injection, and then gradually decreased from 24 h to 72 h (Fig. 5b). As shown in Fig. 5c and d, remarkably, nanoparticles were accumulated in the tumor tissue, and its fluorescence signal was significantly higher than that in the normal tissue. The passive targeting by the nanoparticles was due to EPR effect, while free IR775 could not accumulate in the tumor region post intravenous injection, as previously reported.30 | ||
| Fig. 5 Accumulation of M-Abx and overcoming tumor resistance. (a) Schematic of the established tumor model and therapeutic treatments. (b) Dynamic fluorescence imaging after intravenous injection of IR775@M-Abx. (c–d) NIR fluorescence images and quantification in the major organs and tumor at 10 h post-injection (n = 3), respectively. (e) Relative H2O2 content within the tumor tissues after different treatments (n = 4). (f) Immunofluorescence images of tumor sections stained by HIF-1α (green), VEGF (red), and P-gp (red) after intravenous injection of vehicle, MnO2-HSA, Abx and M-Abx, scale bar = 100 μm. (g–i) Quantification of fluorescence intensity from (f) (n = 3). Data is shown as mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001. | ||
Then, the impact of H2O2 content in the tumor tissues was determined after different treatments. The H2O2 content significantly increased after Abx treatment, but M-Abx could consume the additional H2O2 produced by Abx and keep the content of H2O2 at approximately the level of the control group (Fig. 5e). After 36 h post intravenous injection of Abx, HIF-1α expression was remarkably upregulated, which might be due to the function of increased H2O2 to stabilize HIF-1α.31 In terms of M-Abx treatment, H2O2 could be catalyzed to generate O2 to initiate the oxygen-dependent HIF-1α degradation pathway. As expected, the content of HIF-1α significantly decreased in the M-Abx group compared with that in the Abx group (Fig. 5f and g, respectively).
P-gp and VEGF are downstream genes controlled by HIF-1α, which play vital roles in tumor chemotherapy resistance and metastasis, respectively.32 Paclitaxel is one of the well-known substrates of P-gp expressed on tumor cells.33 After Abx stimulation, there was considerable P-gp expression, which could pump out paclitaxel, inducing acquired resistance to chemotherapy. The decreased P-gp in the M-Abx group indicates enhanced chemotherapy due to improved paclitaxel inside the tumor cells for disrupting the function of microtubulin (Fig. 5f and h). VEGF has been implicated in angiogenesis, tumor growth and metastasis. Similarly, after treatment with M-Abx, the expression of VEGF was significantly suppressed, suggesting the lower tendency of tumor invasion and metastasis (Fig. 5f and i).
3.5 Enhanced therapeutic effect of M-Abx in vivo
To inspect the tumor suppression of M-Abx in vivo, mice bearing 4T1 tumor were randomly divided into four groups, including vehicle, MnO2-HSA, Abx and M-Abx. As shown in Fig. 6a, MnO2-HSA alone did not inhibit the tumor growth during treatment, and about 35% inhibition of tumor growth was achieved after Abx treatment. However, in the M-Abx group, the most optimal therapeutic effect of 71% inhibition was observed (Fig. 6a and Fig. S4†), which might be due to the downregulation of multidrug resistance proteins. During treatment, there was no observable difference in body weight between the M-Abx and Abx groups, indicating that the introduction of the MnO2 structure did not result in additional systemic toxicity (Fig. 6b). | ||
| Fig. 6 Enhanced tumor therapeutic efficiency of M-Abx in 4T1 bearing mice. (a) Tumor growth curves after the indicated treatments. Treatments were performed on day 0, 3, and 7. Data is shown as mean ± SEM (n = 6). (b) Body weight of mice in the different groups. Data is shown as mean ± SD (n = 6). (c) Images of the tumor sections stained by H&E, TUNEL and Ki67; scale bar = 100 μm. The tumor slices for H&E were obtained on day 15 after monitoring the therapeutic efficiency. The tumor sections for the TUNEL assay and Ki67 immunochemistry were collected at 36 h after intravenous injection of the different treatments. (d–e) Quantification of TUNEL mean fluorescence intensity and Ki67 positive cells after different treatments. Data is shown as mean ± SD (n = 3). *p < 0.05; **p < 0.01; and ***p < 0.001. | ||
Similarly, H&E staining of the tumor sections obtained from the mice on day 15 after chemical therapy showed that M-Abx treatment induced significant tumor necrosis areas and cell nucleus dispersion. In contrast, there were scattered destroyed areas after Abx treatment, where the close arrangement of tumor cells with condensed nuclei indicated an insufficient therapeutic effect (Fig. 6c). Furthermore, TUNEL staining was performed to evaluate the apoptotic cells. As shown in Fig. 6c, Abx treatment only resulted in sporadic red fluorescence within the tumor tissues, which indicates its poor therapeutic efficacy. In the M-Abx group, much more apoptotic cells were induced compared with the other three groups (Fig. 6d). We then evaluated the proliferative tumor cells via immunohistochemical staining of Ki67, which is a well-known biomarker for highly proliferative cells. There was no observable difference in the percentage of Ki67 positive cells between the MnO2-HSA and vehicle groups. The Abx group exhibited 37.85% highly proliferative cells, while a smaller quantity of Ki67+ cells survived after M-Abx treatment (Fig. 6c and e, respectively). All these results manifested that oxygen-rich chemotherapy mediated by M-Abx can efficiently relieve acquired resistance to achieve optimal tumor inhibition.
3.6 Inhibition of tumor metastasis by M-Abx in vivo
HIF-1α-related proteins, such as VEGF, have been implicated in tumor invasion and metastasis. As indicated by the above results, the expression of VEGF remarkably decreased after M-Abx treatment (Fig. 4f). Thus, it was worth investigating whether the tendency of tumor metastasis would be suppressed in the M-Abx group. After 14-day therapy, the primary tumors were removed via surgery. The skin tissue surrounding the tumor tissue was also carefully removed to minimize the risk of primary tumor relapse.34Thirty days later, abundant metastatic lesions were observed in the lung tissue in the control and MnO2-HSA groups (Fig. 7a). In addition, oxygen-rich chemotherapy by M-Abx led to much less observable metastatic lesions than Abx chemotherapy alone (Fig. 7b). The H&E sections showed that large areas of metastatic lesions with condensed nuclei distinct from normal tissue were distributed in the lung tissue of all the groups except the M-Abx group (Fig. 7c). The liver is also a vulnerable organ for tumor metastasis. Similarly, there were much fewer metastatic lesions within the liver in the M-Abx group than the other three groups (Fig. 7c).
 | ||
| Fig. 7 Effective inhibition of tumor metastasis by M-Abx. (a) Images of lung tissues collected on day 44 fixed with Bouin's solution. The black circles indicate the metastatic nodules. (b) Quantification of metastatic lesions in the lung tissues (n = 6). (c) H&E slices of the lung tissues from (a), scale bar = 1 mm and images of the H&E sections of the liver, scale bar = 200 μm. (d–e) Survival rate curves (data was analyzed using the log rank test) and survival time of the mice in the different groups after intravenous injection of 4T1 cells. Data is shown as mean ± SD (n = 6). *p < 0.05; **p < 0.01; and ***p < 0.001. | ||
In order to inspect the inhibition of metastasis by M-Abx directly, 4T1 cells were intravenously injected into mice to establish an experimental metastatic model. After the injection of the tumor cells, chemotherapy was performed on day 13, and 21. Unfortunately, Abx did not significantly increase the survival rate of the 4T1 metastatic mice compared with the vehicle group. However, after M-Abx treatment, the survival rate and average survival time significantly improved, which indicates the inspiring optimal therapeutic effect of metastasis suppression (Fig. 7d and e, respectively).
3.7 Toxicity tests of M-Abx in vivo
To evaluate the toxicity of M-Abx during treatment, major tissue sections and serum biochemistry analysis were performed in a separate experiment, where the treatment schedule was the same as the above in vivo therapeutics. Blood and major organs from the mice were carefully collected and analyzed. As shown in Fig. 8a, there was no observable damage to the major organs of the mice (including the heart, liver, spleen, lung and kidney) compared with the vehicle group. Furthermore, the liver function markers including alanine aminotransferase (ALT) and aspartate aminotransferase (AST) indicated that no observable damage to liver function occurred after M-Abx treatment (Fig. 8b and c). The kidney function marker (blood urea nitrogen, BUN) also showed that M-Abx did not induce significant damage to renal function (Fig. 8d). Direct bilirubin (DB), total bilirubin (TB) and the ratio of DB/TB showed no difference in the influence of hemolysis between M-Abx and the other groups (Fig. 8e–g). All these results manifest the safety of oxygen-rich chemotherapy mediated by M-Abx. | ||
| Fig. 8 Toxicity tests of M-Abx in vivo. (a) H&E staining of the major organs of the different groups including the heart, liver, spleen, lung and kidney, scale bar = 100 μm. (b–c) Serum biochemistry data reflecting liver function including ALT and AST. (d) Serum biochemistry data of BUN reflecting kidney function. (e–g) Serum biochemistry data reflecting the hemolysis of the different treatments, including DB, TB and ratio of DB/TB. Data is shown as mean ± SD (n = 6). NS represents non-significance. | ||
4. Discussion
Abraxane® is widely used in the treatment of mTNBC. However, the outcome of traditional Abx chemotherapy is not satisfactory. Many studies indicated that paclitaxel could induce acquired resistance.35–37 However, paclitaxel treatment could stimulate tumor cells to produce H2O2 to stabilize HIF-1α, which would dimerize with HIF-1β, and then upregulate P-gp, VEGF and other resistance-related proteins.10,12,31 Herein, we also confirmed that abundant H2O2 was generated after Abx treatment. As a result, HIF-1α and downstream P-gp were upregulated, indicating the acquired resistance to chemotherapy.Some studies involved combining P-gp inhibitors with chemotherapeutic agents to overcome the acquired resistance.38–41 Unfortunately, most of these studies failed in clinical trials, which might be attributed to their unfavorable pharmacological properties, pharmacokinetic interactions and toxicity.42–44 Instead of applying P-gp inhibitors, we took advantage of M-Abx to realize oxygen-rich chemotherapy to inhibit the expression of P-gp to enhance the therapeutic effects of paclitaxel. The benefits of oxygen-rich chemotherapy covers two aspects. Since sufficient H2O2 only exists in the tumor microenvironment, oxygen generation is tumor-region specific, which is dependent on the reaction between MnO2 and H2O2. The improvement in oxygenation and decrease in H2O2 leads to the degradation of HIF-1α. Subsequently, the expression of resistance-related proteins such as P-gp can be notably inhibited. Moreover, the downregulation of tumor invasion related genes such as VEGF can reduce the risk of systemic metastasis. The efficient control of metastasis progression via oxygen-rich chemotherapy was also confirmed in both spontaneous and experimental metastatic models.
Apart from mTNBC, the oxygen-rich chemotherapeutic strategy is also suitable for other types of tumors. In the CT26 colon tumor model, Abraxane® showed negligible therapeutic efficiency, while M-Abx achieved 50.6% inhibition of tumor growth without increasing systemic toxicity (Fig. S5 and S6,† respectively).
The integration of diagnosis and treatment is an emerging field in tumor therapy. Magnetic resonance imaging (MRI) is widely applied in clinic owing to its safety, noninvasiveness, and potent penetration depth.45 The most common MRI contrast agents are based on Gd3+ and Mn2+-related products for shortening the T1 relaxation time.46 As normal tissues only contain trace H2O2, the reduction reaction of MnO2 into Mn2+ is limited to the tumor region with overexpressed H2O2.47 Therefore, M-Abx is of great potential to achieve tumor imaging for evaluating tumor progression.
It is of great importance that scientific research, particularly for tumor therapy, should be transformed into clinical treatment. Simplification of manufacturing techniques, admirable stability of nanodrugs and the low cost of final products are critical considerations from companies and investors. Therefore, instead of complicated chemical modifications using multiple organic solvents, we developed a facile method to integrate a tumor oxygenation agent with the FDA approved Abraxane®. The final product M-Abx can remain stable in PBS and urea solutions, indicating that ionic bonds and hydrogen bonds might not be the main force mediating the affinity between MnO2 and albumin (Fig. S7 and S8†).48 In addition, M-Abx showed favorable stability during storage at 4 °C (Fig. S9 and S10†). Moreover, the modification process did not result in significant changes in the critical characteristics of the parent Abx, including particle size, content and release profile of paclitaxel, which has great potential for clinical application.
5. Conclusion
In this study, we developed a facile oxidation method to obtain oxygen-rich chemotherapeutic nano-medicine. The fabrication of M-Abx did not change the main characteristics of the parent commercial Abraxane®, which resulted in favorable stability and therapeutic effectiveness. After accumulation within the tumor region, M-Abx could catalyze the overexpressed H2O2 to generate O2 for tumor oxygenation to induce oxygen-dependent HIF-1α degradation and further downregulate downstream proteins, including P-gp and VEGF. Overall, oxygen-rich chemotherapy was successfully realized by M-Abx to efficiently overcome acquired resistance and systemic metastasis against mTNBC.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This article was supported by The National Key Research and Development Program of China (No. 2017YFA0205400) and NSFC (Natural Science Foundation of China) (No. 81603043, 81473146). This project is also supported by the Open Fund of State Key Laboratory of Natural Medicines (SKLNMKF201608) and Central Fundamental Research Funds for the Central Universities.Notes and references
- R. Dent, M. Trudeau, K. I. Pritchard, W. M. Hanna, H. K. Kahn, C. A. Sawka, L. A. Lickley, E. Rawlinson, P. Sun and S. A. Narod, Clin. Cancer Res., 2007, 13, 4429–4434 CrossRef PubMed.
- K. R. Bauer, M. Brown, R. D. Cress, C. A. Parise and V. Caggiano, Cancer, 2007, 109, 1721–1728 CrossRef PubMed.
- S. Cleator, W. Heller and R. C. Coombes, Lancet Oncol., 2007, 8, 235–244 CrossRef PubMed.
- C. Denkert, C. Liedtke, A. Tutt and G. von Minckwitz, Lancet, 2017, 389, 2430–2442 CrossRef CAS.
- L. Gianni, M. Mansutti, A. Anton, L. Calvo, G. Bisagni, B. Bermejo, V. Semiglazov, M. Thill, J. I. Chacon, A. Chan, S. Morales, I. Alvarez, A. Plazaola, M. Zambetti, A. D. Redfern, C. Dittrich, R. A. Dent, D. Magazzu’, P. Valagussa and I. Tusquets, J. Clin. Oncol., 2016, 34, 502 Search PubMed.
- T. E. Stinchcombe, M. A. Socinski, C. M. Walko, B. H. O'Neil, F. A. Collichio, A. Ivanova, H. Mu, M. J. Hawkins, R. M. Goldberg, C. Lindley and E. C. Dees, Cancer Chemother. Pharmacol., 2007, 60, 759–766 CrossRef CAS PubMed.
- M. R. Green, G. M. Manikhas, S. Orlov, B. Afanasyev, A. M. Makhson, P. Bhar and M. J. Hawkins, Ann. Oncol., 2006, 17, 1263–1268 CrossRef CAS PubMed.
- R. B. Tang, F. H. Yan, W. M. Chai, W. Huang, Y. N. Fu, G. Y. Yang and K. M. Chen, J. Synchrotron Radiat., 2015, 22, 1263–1267 CrossRef CAS PubMed.
- R. Z. Yusuf, Z. Duan, D. E. Lamendola, R. T. Penson and M. V. Seiden, Curr. Cancer Drug Targets, 2003, 3, 1–19 CrossRef CAS PubMed.
- J. Alexandre, Y. M. Hu, W. Q. Lu, H. Pelicano and P. Huang, Cancer Res., 2007, 67, 3512–3517 CrossRef CAS PubMed.
- T. Hadzic, N. Aykin-Burns, Y. M. Zhu, M. C. Coleman, K. Leick, G. M. Jacobson and D. R. Spitz, Free Radical Biol. Med., 2010, 48, 1024–1033 CrossRef CAS PubMed.
- J. M. Oh, Y. K. Ryu, J. S. Lim and E. Y. Moon, Biomol. Ther., 2010, 18, 145–151 CrossRef CAS.
- M. W. Dewhirst, Y. Cao and B. Moeller, Nat. Rev. Cancer, 2008, 8, 425–437 CrossRef CAS PubMed.
- A. Giaccia, B. G. Siim and R. S. Johnson, Nat. Rev. Drug Discovery, 2003, 2, 803–811 CrossRef CAS PubMed.
- G. L. Semenza, Trends Mol. Med., 2002, 8, S62–S67 CrossRef CAS PubMed.
- K. M. Comerford, T. J. Wallace, J. Karhausen, N. A. Louis, M. C. Montalto and S. P. Colgan, Cancer Res., 2002, 62, 3387–3394 CAS.
- P. D. W. Eckford and F. J. Sharom, Chem. Rev., 2009, 109, 2989–3011 CrossRef CAS PubMed.
- G. Arancia, A. Molinari, A. Calcabrini, S. Meschini and M. Cianfriglia, Marcello Malpighi Symp. Ser., 2001, 7, 59–68 Search PubMed.
- N. Ferrara, H. P. Gerber and J. LeCouter, Nat. Med., 2003, 9, 669–676 CrossRef CAS PubMed.
- P. Carmeliet, Y. Dor, J. M. Herbert, D. Fukumura, K. Brusselmans, M. Dewerchin, M. Neeman, F. Bono, R. Abramovitch, P. Maxwell, C. J. Koch, P. Ratcliffe, L. Moons, R. K. Jain, D. Collen and E. Keshert, Nature, 1998, 395, 525–525 CrossRef CAS.
- P. Prasad, C. R. Gordijo, A. Z. Abbasi, A. Maeda, A. Ip, A. M. Rauth, R. S. DaCosta and X. Y. Wu, ACS Nano, 2014, 8, 3202–3212 CrossRef CAS PubMed.
- Q. Chen, X. D. Liu, J. W. Chen, J. F. Zeng, Z. P. Cheng and Z. Liu, Adv. Mater., 2015, 27, 6820–6827 CrossRef CAS PubMed.
- Q. Chen, L. Z. Feng, J. J. Liu, W. W. Zhu, Z. L. Dong, Y. F. Wu and Z. Liu, Adv. Mater., 2016, 28, 7129–7136 CrossRef CAS PubMed.
- G. B. Yang, L. G. Xu, Y. Chao, J. Xu, X. Q. Sun, Y. F. Wu, R. Peng and Z. Liu, Nat. Commun., 2017, 8, 902 CrossRef PubMed.
- W. W. Zhu, Z. L. Dong, T. T. Fu, J. J. Liu, Q. Chen, Y. G. Li, R. Zhu, L. G. Xu and Z. Liu, Adv. Funct. Mater., 2016, 26, 5490–5498 CrossRef CAS.
- L. T. Meng, Y. L. Cheng, S. J. Gan, Z. C. Zhang, X. N. Tong, L. Xu, X. Jiang, Y. S. Zhu, J. H. Wu, A. Yuan and Y. Q. Hu, Mol. Pharmaceutics, 2018, 15, 447–457 CrossRef CAS PubMed.
- P. Caironi, G. Tognoni, S. Masson, R. Fumagalli, A. Pesenti, M. Romero, C. Fanizza, L. Caspani, S. Faenza, G. Grasselli, G. Iapichino, M. Antonelli, V. Parrini, G. Fiore, R. Latini, L. Gattinoni and A. S. Investigators, N. Engl. J. Med., 2014, 370, 1412–1421 CrossRef CAS PubMed.
- J. Uriz, P. Gines, A. Cardenas, P. Sort, W. Jimenez, J. M. Salmeron, R. Bataller, A. Mas, M. Navasa, V. Arroyo and J. Rodes, J. Hepatol., 2000, 33, 43–48 CrossRef CAS PubMed.
- L. Meng, Y. Cheng, X. Tong, S. Gan, Y. Ding, Y. Zhang, C. Wang, L. Xu, Y. Zhu, J. Wu, Y. Hu and A. Yuan, ACS Nano, 2018, 12, 8308–8322 CrossRef CAS PubMed.
- X. L. Tang, G. J. Wang, R. J. Shi, K. Jiang, L. T. Meng, H. Ren, J. H. Wu and Y. Q. Hu, Drug Delivery, 2016, 23, 2686–2696 CAS.
- D. Samanta, D. M. Gilkes, P. Chaturvedi, L. S. Xiang and G. L. Semenza, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E5429–E5438 CrossRef CAS PubMed.
- G. L. Semenza, Nat. Rev. Cancer, 2003, 3, 721–732 CrossRef CAS PubMed.
- S. Callies, D. P. de Alwis, A. Harris, P. Vasey, J. H. Beijnen, J. H. Schellens, M. Burgess and L. Aarons, Br. J. Clin. Pharmacol., 2003, 56, 46–56 CrossRef CAS PubMed.
- P. B. Gupta, C. Kuperwasser, J. P. Brunet, S. Ramaswamy, W. L. Kuo, J. W. Gray, S. P. Naber and R. A. Weinberg, Nat. Genet., 2005, 37, 1047–1054 CrossRef CAS PubMed.
- P. Liu, I. S. Kumar, S. Brown, V. Kannappan, P. E. Tawari, J. Z. Tang, W. Jiang, A. L. Armesilla, J. L. Darling and W. Wang, Br. J. Cancer, 2013, 109, 1876–1885 CrossRef CAS PubMed.
- C. Swanton, M. Marani, O. Pardo, P. H. Warne, G. Kelly, E. Sahai, F. Elustondo, J. Chang, J. Temple, A. A. Ahmed, J. D. Brenton, J. Downward and B. Nicke, Cancer Cell, 2007, 11, 498–512 CrossRef CAS PubMed.
- E. K. Rowinsky and R. C. Donehower, N. Engl. J. Med., 1995, 332, 1004–1014 CrossRef CAS PubMed.
- F. H. Wang, D. R. Zhang, Q. Zhang, Y. X. Chen, D. D. Zheng, L. L. Hao, C. X. Duan, L. J. Jia, G. P. Liu and Y. Liu, Biomaterials, 2011, 32, 9444–9456 CrossRef CAS PubMed.
- X. Y. Wang, Y. H. Chen, F. Z. Dahmani, L. F. Yin, J. P. Zhou and J. Yao, Biomaterials, 2014, 35, 7654–7665 CrossRef CAS PubMed.
- H. Thomas and H. M. Coley, Cancer Control, 2003, 10, 159–165 CrossRef PubMed.
- A. G. Assanhou, W. Y. Li, L. Zhang, L. J. Xue, L. Y. Kong, H. B. Sun, R. Mo and C. Zhang, Biomaterials, 2015, 73, 284–295 CrossRef CAS PubMed.
- G. Szakacs, J. K. Paterson, J. A. Ludwig, C. Booth-Genthe and M. M. Gottesman, Nat. Rev. Drug Discovery, 2006, 5, 219–234 CrossRef CAS PubMed.
- M. V. Seiden, K. D. Swenerton, U. Matulonis, S. Campos, P. Rose, G. Batist, E. Ette, V. Garg, A. Fuller, M. W. Harding and D. Charpentier, Gynecol. Oncol., 2002, 86, 302–310 CrossRef CAS PubMed.
- P. M. Fracasso, M. F. Brady, D. H. Moore, J. L. Walker, P. G. Rose, L. Letvak, T. M. Grogan and W. P. McGuire, J. Clin. Oncol., 2001, 19, 2975–2982 CrossRef CAS PubMed.
- R. G. H. Beets-Tan, G. L. Beets, R. F. A. Vliegen, A. G. H. Kessels, H. Van Boven, A. De Bruine, M. F. von Meyenfeldt, C. G. M. I. Baeten and J. M. A. van Engelshoven, Lancet, 2001, 357, 497–504 CrossRef CAS.
- H. B. Na, I. C. Song and T. Hyeon, Adv. Mater., 2009, 21, 2133–2148 CrossRef CAS.
- B. Frei, Y. Yamamoto, D. Niclas and B. N. Ames, Anal. Biochem., 1988, 175, 120–130 CrossRef CAS PubMed.
- M. C. Stumpe and H. Grubmuller, J. Phys. Chem. B, 2007, 111, 6220–6228 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8bm00753e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |