
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The application of surface enhanced Raman scattering to the detection of asphaltic petroleum in sediment extracts: deconvolving three component-mixtures using look-up tables of entire surface enhanced Raman spectra†
Stephen A.
Bowden
 * and
Colin W.
Taylor
* and
Colin W.
Taylor
School of Geosciences, University of Aberdeen, Aberdeen, AB243UE, UK. E-mail: s.a.bowden@abdn.ac.uk
First published on 11th November 2019
Abstract
The detection of asphaltic petroleum by surface enhanced Raman scattering (SERS) is uncomplicated, except in instances where the petroleum has been mixed with other components that also exhibit a strong Raman effect. Such a situation is common where, for example, petroleum is mixed with other naturally occurring materials in sedimentary environments. Sedimentary organic matter in deserts, on beaches, on continental shelves and in the deep sea may contain a variety of compound types that includes pigments and humic compounds, all of which are also amenable to surface enhanced Raman scattering, making the detection of asphaltic petroleum by SERS in these natural environments complicated. However, for a beach-sediment comprising sediment weathered from exhumed petroleum reservoirs and source rocks (naturally occurring sources of petroleum) it can be shown that the asphaltic component of a surface enhanced Raman spectra can be found by deconvolving asphaltene, humic acid and pigments using entire spectra. This is achieved by finding matches on look-up tables comprising entire spectra with varied contributions from different compounds. In using the entire spectra, subjectivity introduced by fitting individual curves to match multiple Raman bands is reduced, minimising the consequences of what can otherwise be a process that involves arbitrary decision making. Using this approach it can be shown that a SERS-assay of asphaltic materials in sediments from a foreshore in Northern Scotland can produce results that benchmark reasonably against standard methods, and data that yields interpretations of the natural environment that are consistent with that derived from comparator GC-MS data.
1 Introduction
The detection of petroleum components in sedimentary organic matter is straightforward when petroleum forms the main component, or when well-established analytical techniques for the isolation and separation of target analytes exist,1,2 as is the case for petroleum biomarker analysis by solvent extraction and GC-MS analysis. However not all petroleum components within sedimentary organic matter are amenable to GC-MS. This is particularly so for asphaltic materials which can form from the weathering of petroleum within sedimentary environments, via microbial degradation of the most labile and volatile compounds such as petroleum biomarkers.3 This causes a reduction in the proportion of volatile compounds and a selective enrichment in the asphaltic component of petroleum.‡In addition to asphaltic petroleum there are other compound-classes that organic geochemists might include in a catch-all group of organic components that remain after the GC-MS-amenable compounds have been isolated or removed. One such term for these compounds is the molecularly uncharacterised component of organic matter.4 Clearly what is uncharacterisable depends partly on the technological capabilities available as well as recognition of what is present. Asphaltene, asphalt and related materials are the component of petroleum that is least volatile and least soluble.5 Techniques for the characterisation of asphaltene within petroleum based on FT-ICRMS now permit a far more detailed molecular characterisation of this fraction,6 making the term “uncharacterised component of organic matter” less meaningful. Other large fractions of sedimentary organic matter include the humic acids,2 that are also now easier to characterise at the molecular level with the development of new technologies such as FT-ICRMS.
Pigments are a component of sedimentary organic matter that is also outside of the analytical window provided by GC-MS, although when fresh samples are well-preserved pigments are easily identified by spectroscopic or liquid chromatographic methods. In favourable settings these otherwise readily oxidised compounds may persist for some time, but even in this case pigments (particularly carotenoids) undergo intricate degradation pathways in which functional groups or sites of unsaturation react to convert a single precursor into many lower carbon number homologues or slightly altered structural derivatives.7,8 The preservation of pigments appears to be highly dependent on depositional environment, with high concentrations of the most common pigments not being routinely preserved over long geological-durations.9 None the less, scytonemin a pyrrole-based pigment, for example, persists in arctic regolith and may even be transported to aquatic environments and is stable within sedimentary environments.10 Thus, carotenoid pigments may be present in sedimentary environments due to their ubiquity whilst other pigments may persist due to their recalcitrance.
Simply applied surface enhanced Raman scattering or SERS (simple in the sense that it is un-hyphenated) is known to detect low masses of pigments,11 humic acid12 and asphaltic petroleum13 within the solvent extracts of sedimentary organic matter. This makes SERS a good method for situations where a high throughput is needed, low masses are expected or when a proximal analysis is required rather than specific molecular data. Often working with low masses, published accounts of the application of these pathfinder assays have tended to focus on end-member cases where identification and interpretation of spectra is simple – for example the weathering products of highly pigmented endolithic microbes, soil or PAH within seawater.14,15 These assays are very powerful in the sense that for certain compound types these methods are highly sensitive, whilst having minimal infrastructure requirements making them suitable for point of need or remote deployment via handheld units, or stand-alone modules on boats, space vehicles and planetary landers.14,16,17
For SERS, the combined high selectivity and high sensitivity are highly desirable, as these two aspects reduce potential interferences from compounds that do not exhibit the Raman effect whilst maximising signal from the analyte. However, because of the prevalence of both humic, pigmented and asphaltic organic matter in the natural environment the potential exists for one or more of these compound types to interfere with the SERS-analysis of another.
Here we present the application of SERS to detecting naturally occurring asphaltic petroleum in beach sediments and demonstrate a processing algorithm that overcomes some of the problems presented by real world samples that also contain interfering humic acid and biological pigments. In the following work we compare the method to comparator methods for measuring asphaltic petroleum, and within a natural science context compare the SERS assay of asphaltic petroleum to other data types more typically used to measure petroleum in beach sediments.
2 Experimental
2.1 Field site
The site, as shown in Fig. 1a–c, comprises a bay with cliffs and a foreshore separated by a manmade jetty. On the eastern side of the jetty is a beach made of shingle weathered from the flagstone exposed in the cliff. The flagstone is a potential petroleum source rock (Fig. 1d and e); a rock with a sufficiently high organic carbon content that if exposed to the correct thermal conditions generates petroleum.18 The shingle weathered from the rock retains a high proportion of solvent extractable bitumen (Fig. 1e and Table 1), and forms flat oblate shapes a few mm thick. | ||
| Fig. 1 (a) Brough in Northern Scotland, UK. (b) Map showing the two sections of the bay with different rocks exposed in the cliffs. (c) Photo of site, looking west, showing the wave cut foreshore and cliffs. (d) Flagstone and (e) shingle eroded from the flagstone. (f) Petroleum-baring sandstones and (g) sand eroded from the sandstone. | ||
| Sediment | XRF % oxide | EOM, mg g−1 | SARA | SERS | SERS | Sterane petroleum biomarkers | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sieve size | MgO | Al2O3 | SiO2 | Asphaltene | % Asphaltene | n | mg g−1 sed | n | ng g−1 sed | |||||||||
| mm | %![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
% | mg g−1 | Mean | Max | Min | Mean | Max | Min | Steranes | Diasteranes | |||||||
| a EOM = extractable organic matter, SARA = saturate aromatic, resin, asphaltene, steranes = sum of C29 regular sterane homologues, diasteranes = sum of all of the C29 diasteranes. | ||||||||||||||||||
| Flagstone | 1.5 | 3.3 | 12 | 0.46 | 8.7 | 0.04 | 5.5 | 7.8 | 2.7 | 5 | 0.025 | 0.04 | 0.01 | 5 | 3.66 | 7.8 | ||
| Coarse | >1 | 82 | 1.3 | 2.9 | 13 | 0.12 | 35 | 0.04 | 39 | 38 | 2 | 0.04 | 0.04 | 2 | 0.16 | 0.9 | ||
| Fine | <1 | 18 | 1.4 | 2.9 | 14 | 0.17 | 63 | 0.11 | 45 | 39 | 2 | 0.08 | 0.07 | 2 | 0.05 | 0.3 | ||
| Clean | 0.38 | 8.6 | 0.03 | 10 | 11 | 7.7 | 4 | 0.038 | 0.04 | 0.03 | 4 | |||||||
| Sandstone | 0.8 | 3.2 | 18 | 1.19 | 5.1 | 0.06 | 11 | 2.1 | 2 | 0.13 | 0.03 | 2 | 8.83 | 26 | ||||
| Coarse | >0.5 | 39 | 0.9 | 1.0 | 9 | 0.36 | 71 | 0.26 | 73 | 72 | 2 | 0.36 | 0.26 | 2 | 0.07 | 0.3 | ||
| Fine | <0.5 | 61 | 0.8 | 1.0 | 12 | 0.07 | 30 | 0.02 | 43 | 23 | 2 | 0.03 | 0.001 | 2 | 0.05 | 0.3 | ||
| Clean | 0.47 | 46 | 0.21 | 52 | 34 | 0.28 | 0.28 | |||||||||||
| Coarse | >0.5 | 61 | 1.1 | 1.4 | 9 | 0.13 | 37 | 0.05 | 14 | 8.4 | 2 | 0.02 | 0.01 | 2 | 0.13 | 0.6 | ||
| Fine | <0.5 | 39 | 0.9 | 0.9 | 13 | 0.12 | 28 | 0.03 | 22 | 2.6 | 2 | 0.03 | 0.003 | 2 | 0.05 | 0.3 | ||
| Clean | 0.38 | 34 | 0.13 | 35 | 14 | 2 | 0.33 | 0.05 | 2 | |||||||||
On the western side of the jetty the sand on the foreshore comprises sand weathered from a cliff of petroleum-impregnated sandstone (Fig. 1f and g). Under subsurface conditions, were the oil within the sandstone saturated with gas, it would be considered analogous to a petroleum reservoir.19 At surface conditions this is not the case and the petroleum is never a free-flowing liquid phase (even during summer), instead it is a solid phase within the sandstone or bound to the surfaces of sand formed from the weathering of the petroleum impregnated sandstone. For all samples, from the perspective of a SERS-assay, a number of analytical interferences are naturally present; endoliths within the sandstone (visible as green and purple colouration beneath the surface of the sandstones, Fig. 1g), fragments of other plants and marine organisms within the beach sand and shingle. As most samples are not pristine but weathered, pure pigment (carotenoid or tetra-pyrrole) would not be expected to be present and instead a range of structural-derivatives would be anticipated as described above (see Wilson et al.11 for examples of how this may broaden SERS-bands).
2.2 Spike study and additional samples
In addition to the samples from Brough, a spike study was also performed using a deep marine sediment sample from the South China Sea.20 This study was performed to evaluate the deconvolution procedure in a different sample set (a deep as opposed to shallow marine sample), and to evaluate the deconvolution procedure under controlled conditions. The sample of deep marine mud came from a water depth of ∼3000 meters and comprises a fine-grained mud (sample IODP 368 U1501-C 1-1H, 0–5 cm).20 The sample was homogenised and split into aliquots. The marine mud and samples of homogenised Californian tar sand (sample used in Alabi et al.13) were mixed to produce the samples shown in Table 2 with the shown masses of asphalt and sedimentary organic matter. One sub-group comprises only petroleum-rich samples while the other comprises a petroleum-rich series mixed with marine mud and solvent extractable sedimentary organic matter. Additional samples were used to provide a natural science context and these are listed in Table 3.| Asphaltene, μg | Deep marine mud extract, μg | |
|---|---|---|
| a Standard of Californian tar sand listed in Table 3. b Spike was added to extract of marine mud listed in Table 3. | ||
| Procedural blank | 0 | 0 |
| Standarda | 2 | 0 |
| Standard | 3 | 0 |
| Standard | 5 | 0 |
| Spiked sampleb | 0.8 | 7.5 |
| Spiked sample | 2.4 | 7.5 |
| Spiked sample | 4.1 | 7.5 |
| Spiked sample | 5.4 | 7.5 |
| Sample | Purpose | Sample description | Diasterane | Asphaltene | Reference in source | Source |
|---|---|---|---|---|---|---|
| Extract marine mud | Spike study | Solvent extract, deep marine mud | nd | nd | IODP 368 U1501-B-ML | Larsen et al.20 |
| Marine mud | Natural context (Fig. 6) | Deep marine mud | 0.044 ng g−1 | 2.7 mg g−1 | IODP 368 U1501-C1-1H, 0–5 cm | Larsen et al.20 |
| Athabasca tar sand | Natural context (Fig. 6) | Variably weathered tar sand from Athabasca | 0.20 ng g−1 | 48 mg g−1 | CAN 1 | Bowden21 |
| Athabasca tar sand | Natural context (Fig. 6) | Variably weathered tar sand from Athabasca | 0.19 ng g−1 | 2.7 mg g−1 | CAN 9 | Bowden21 |
| Californian tar sand | Reference asphaltic petroleum & spike study | Tar sand from California | — | — | CAT-1 | Alabi et al.13 |
| Wearing coarse sphalt | Natural context (Fig. 6) | Sample of weathered wearing coarse asphalt | 0.006 ng g−1 | 161 mg g−1 | BOD-A-2017 | — |
2.3 Preparation and analysis
After collection sediment and rock samples were freeze-dried before weighing and analysis. Samples of sediment from the foreshore at Brough had large pieces of extraneous organic matter removed and were further cleaned prior to sonicating the sediment in 1:1 dichloromethane/methanol (DCM/MeOH) v/v for 3 minutes. Fine and coarse sized factions of the sediment were obtained by sieving the sediment using a 1 mm size mesh for the flagstone and 0.6 mm size mesh for the sandstone (mesh seizes correspond to minimum dimensions; ∼1.2 mm for laminations in flagstone and 0.6 mm for fine sand in the sandstone).
All samples were crushed and subjected to solvent extraction using soxhlet apparatus (5–25 g of sediment or rock was extracted in dichloromethane/methanol 93:7 v/v for 48 h). The extract was split into asphaltene and maltene fractions by eluting the asphaltene fraction with an excess of n-heptane, prior to separation using silica-gel flash column chromatography (silica column; hexane for saturated fraction; 3:1 v/v hexane/dichloromethane for aromatics fraction; 2:1 v/v dichloromethane/methane for polar fraction).22,23 To provide a point of reference for the general character of the samples by a complimentary method the resulting saturate fraction was analysed by gas chromatography-mass spectrometry (GC-MS). Aliquots of unseparated extracts were analysed by SERS.
2.4 GC-MS analysis
GC-MS was performed using a 6890N Network GC system connected to a 5975 inert mass selective detector. A PTV injector (300 °C) operating in splitless mode was used and the GC temperature program was; 60 °C to 120 °C at 20 °C min−1 then from 120 °C to 290 °C at 4 °C min−1. The column was Greyhound GC-5 (an HP-5 equivalent phase; 30 m length, 250 μm ID and 0.25 μm film thickness). The MS was operated in sim mode (less than 20 ions with a dwell time less than 40 ms). Compounds were identified by reference to well-characterized samples of bitumen. Inputs for hopanoid biomarker chemometric parameters were measured using peak areas of compounds on the m/z 189 and 191 ion chromatogram. Quantification of steranes and diasteranes was performed relative to an internal standard of 5β-cholane and the m/z 217 and 259 ion chromatograms for steranes and diasteranes.2.5 Surface enhanced Raman spectroscopy
Following the method presented in Alabi et al.13 the extract was dissolved in DCM and 4–8 μl of it introduced in small increments to the sample-wells of a gold-coated SERS-substrate. Introducing samples as small increments allowed the solvent to evaporate under ambient conditions until the entire volume had been loaded. When the solvent had evaporated 4 μl of dilute nitric acid (1 M concentration) was dispensed into the pit and allowed to stand for ∼2–3 minutes. The aqueous phase remaining within the pit was then removed to aid focusing of the microscope on the base of the pit. Raman spectroscopic measurements were performed using a BWTek i-Raman Pro fitted with a 532 nm light source and mounted on 20× video-microscope. Spectra were collected by accumulating 400 spectra of 300 ms duration and spectral acquisition was in the range 200–2000 cm−1, with the region 1200 to 1800 cm−1 used for deconvolution. Laser spot size was approximately 2–4 μm and laser power was 40–60% (<13 mW delivered to the sample). Quantification of asphaltene was performed relative to the asphaltic petroleum obtained from a reference sample of Californian tar sand. Calibration was performed for each batch of samples, typically using standards that doubled in concentration.For both calibration-standards and extracts of sedimentary organic matter, care must be taken to not overload the surface of a SERS-substrate. This is for two reasons: (1) asphaltene is opaque and attenuates both the light transferred to a substrate-surface and the transmission of a SERS-signal to a detector.13 (2) More generally extracts of sedimentary organic matter contain a mixture of SERS-components with varied solubility and thus the potential exists for one compound type to preferentially coat the surface. Because the greater part of a SERS-signal is generated by interaction between analyte and substrate within a few nanometres of a surface, the potential exists for the compounds that first deposit from a solution to disproportionately contribute to a SERS-signal due to a greater enhancement.24 Both problems are mitigated by keeping solutions dilute, the total mass of analyte low and the evaporation of solvent rapid during substrate-loading.
2.6 Processing algorithm
Surface enhanced Raman spectra were deconvolved using the procedure illustrated in Fig. 2. The procedure is automated and comprises 4 stages to deduce the relative contributions of three end-member components to a surface enhanced Raman spectra. In the first stage (Fig. 2a) basic processing removes a baseline and resamples data to a constant frequency of measurement (in this case bin sizes of 1 cm−1). In the second stage (Fig. 2b) a “look-up table” is calculated. The look-up table contains surface enhanced Raman spectra that are a mix of two end member components (in this case humic acids and asphaltene taken from Alabi et al.13). The mix of the two components that best matches the measured spectra is identified. In the third stage (Fig. 2c) a new look-up table is calculated from the best matching spectra from stage 4 (a mixture of humic acid and asphaltene) and the third component (in this case a composite SERS-spectra of ββ-carotene and c-phycocyanin – spectra taken from Bowden et al.14). The best match to the original measured surface enhanced Raman spectra is then identified. The data retuned includes the percentage of the three component spectra (asphaltene, humic acid and pigment) and the goodness of fit of the modelled surface enhanced Raman spectra to the original. The key metric returned is a peak height for the so-called G-band of the asphaltene spectra13 between 1570–1580 cm−1. Example processing scripts (written in R), examples of returned data and data for the component spectra are provided in ESI.†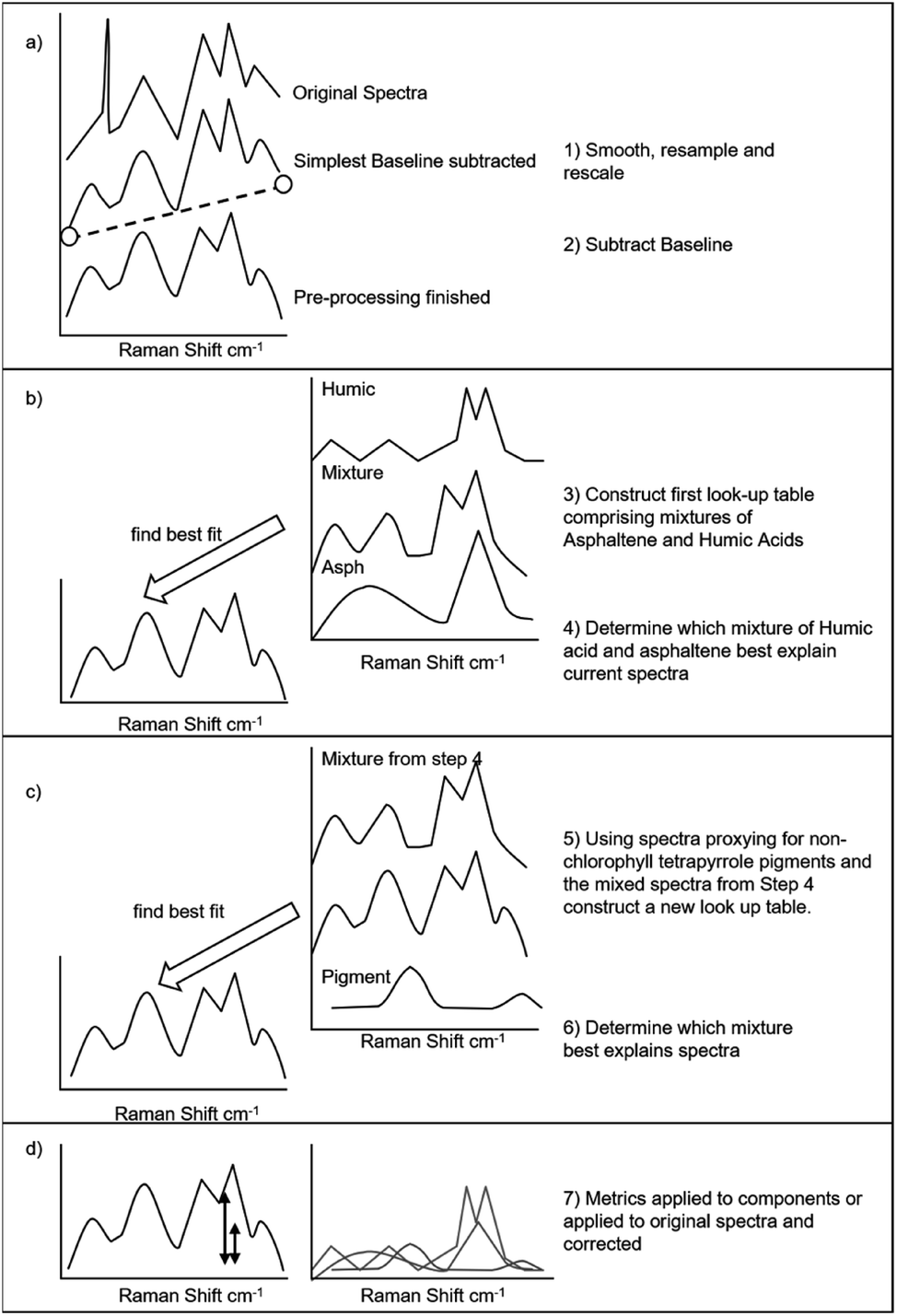 | ||
| Fig. 2 Algorithm used to deconvolve the components of a surface enhanced Raman spectra using look-up tables and entire spectra. (a) During pre-processing the data is smoothed, resampled to set resolution and a baseline subtracted. (b) The mixture of asphaltene and humic organic matter is determined. (c) The mixture of pigment and humic and asphaltic organic matter determined. (d) Metrics returned and quantification performed. | ||
3 Results and discussion
3.1 SERS-spectra and ion chromatograms
The solvent extracts of the largest clasts of the flagstone-shingle collected on the foreshore have surface enhanced Raman spectra with a strong Raman bands around 1300 and 1580 cm−1 (Fig. 3a) similar to the D and G bands seen in kerogen25,26 and previously seen SERS-spectra of asphaltic petroleum.13,14 This is consistent with previous work describing the presence of petroleum source rocks and oil-baring sandstones at Brough.19 The ion chromatograms (Fig. 3b) also contain a well-resolved homologous series of hopanes with structural configurations typically found in oil (e.g. “oil-window” biomarkers – where “oil window” refers to a level of thermal alteration sufficient to generate oil from sedimentary organic matter). Despite features within the surface enhanced Raman spectra that might indicate humic acids, such as the bands at 1400 and 1260 cm−1, by visual inspection of the SERS-spectra alone it would be determined that a high proportion of asphaltic petroleum was present in the flagstone shingle. | ||
| Fig. 3 Surface enhanced Raman spectra for large (coarse) clasts of (a) flagstone and (b) petroleum-baring sandstone. Measurements are show as open circles, asphaltene spectra in brown, humic organic matter in green and β,β-carotene and c-phycocyanin in purple. Spectra for asphaltene and humic organic matter are from Alabi et al.13 The 191 m/z ion chromatograms for (c) flagstone and (d) petroleum baring sandstone. Peaks are labelled for C27 trisnorhopane (Ts); C27 trisnorneohopane (Tm); C29 and C30 14α,17β (H) hopane (C29 and C30 αβ); C31 and C32 14 α,17β (H) 22S & 22R hopanes; C30 diploptene. Inserts show the M − 1 ion chromatogram for diploptene (m/z 419) and a diagnostic ion corresponding the C/D ring fragment (m/z 189). | ||
The surface enhanced Raman spectra of the clasts of sandstone (weathered fragments of petroleum-reservoir present on the foreshore) have a strong similarity to those of the flagstone, but with distinct spectral features that include bands at 1400 and 1260 cm−1, as well as clear bands centred about 1600 cm−1. The bands at ∼1260, ∼1400 cm−1 and two close bands centred about 1600 cm−1 are spectral features reported in humic organic matter.12,13 Thus SERS-spectra exhibit characteristics of asphaltic petroleum, and also humic organic matter (organic matter found in sediments, but that has not been significantly heated or buried). The GC-MS ion chromatograms suggest a similar mixing of petroleum and non-petroleum organic matter as they show that the sandstone extract, in addition to the hopanes common in petroleum, also contains diploptene – a biological hopanoid commonly found in microbes and lower plants.2
A crucial physical difference between the flagstone and sandstone clasts, that impacts their susceptibility to chemical contamination by extraneous sources, is the greater porosity of the sandstone that makes it more habitable to pigmented micro-organisms such as endoliths, as well as more permeable to water.27 Previous studies have used surface enhanced Raman scattering to measure endolithic-pigments within clasts of rock,11 and also used GC-MS measurements of the ratio of diploptene to petrogenic hopanes as a chemometric proxy for the weathering of petroleum-baring sediment.28 Thus, the differences between the SERS-spectra and ion chromatograms of the flagstone and sandstones is consistent with the history and context of the samples (with the latter type of sample being permeable and open to infiltration by present day organic matter).
3.2 Deconvolution and quantification
The deconvolution achieved using the processing algorithm detailed in Fig. 2 is graphically illustrated in Fig. 4a. After applying this procedure, the flagstone was found to have a relatively low proportion of pigments (5%) such as carotenoids and c-phycocyanin, a higher proportion of humic acid (10%) with the greatest contribution being from asphaltic petroleum (85%). In general, all petroleum-baring rock and sediment from Brough has a similar proportion of asphaltic petroleum (>75%), with the sandstone samples generally having higher proportions of humic acid, and organic matter removed from the surfaces of the samples having higher proportions of pigments (Fig. 4b). This is consistent with the sand being mixed with plant organic matter and sample-surfaces being colonised by endolithic microbes (the green colouration in Fig. 1g). | ||
| Fig. 4 (a) Relative contributions of three SERS-active compound-types to the flagstone shingle; asphaltene (brown), humic organic matter (green), ββ-carotene and c-phycocyanin (blue) and coarse shingle (black). (b) Plot of the calculated contributions to the surface enhanced Raman spectra of samples from the Brough site and samples of deep marine mud spiked with asphalt (see Table 2). Squares = data from Brough (yellow = sandstone samples, maroon = flagstone, purple = extract cleaned from surface of samples); circles = deep marine mud spiked with asphalt (blue = spike 0.8 μg, maroon = spike 2.4–4.1 μg, red = 5.4 μg). | ||
The variably spiked samples of deep-sea marine mud array between the asphalt and pigment corner of the ternary diagram as opposed to the humic acid corner, reflecting the deep marine mud being less affected by plant organic matter than the Brough samples that are close to shore. The spiked-marine mud broadly arrays as expected with the samples with the 3.8 and 5.2 μg spikes of asphalt arraying towards the asphaltene corner (red and maroon samples in Fig. 4b).
The importance of applying the deconvolution procedure can be seen in the differences in the peak height at 1580 cm−1 for the corrected and uncorrected data (Fig. 5a and b); the spectra that have not been deconvolved plot significantly off-trend from their asphalt counterparts. The error is approximately ∼25%, which is within the variance seen for repeat measurements on tar-sand samples that have not been mixed with the interfering compounds found within the deep marine mud.
 | ||
| Fig. 5 Comparison between the detector response (peak height) for (a) measured surface enhanced Raman spectra and (b) the surface enhanced Raman spectra corrected for the relative contribution of asphaltene, humic organic matter and pigments (height attributable to the G-peak at 1599 cm−1). Triangles = samples spiked with asphaltic petroleum, circles = tar sand. | ||
It is crucial to recognize that while the quantification is functionally accurate for measuring asphalt within samples, it is not necessarily accurate for measuring pigments and humic acid. While there is a general consistency in data, e.g. the coastal samples have the highest contribution of humic acid and plant organic matter and the deep marine muds have the least, the extraction and sampling procedure are not optimised for analysing petroleum. Specifically, the use of heat to dry extracts, the use of chlorinated solvent and the low amount of methanol in the extracting solvent would not favour the preservation of easily oxidised pigments or the solvation of humic acids.28 A limited amount of environmental context is evident in the deconvolved data for pigments and humic acids, but this is contextual information (e.g. the samples contain these compound types) and not quantitative data (need not be reflective of quantities in the original samples but only that presented to the detector).
3.3 Quantification within a materials testing context
Asphaltic petroleum can be thought of in two contexts; the percentage of asphaltene in oil (e.g. ASTM D4124 (ref. 22) and ASTM D3279 (ref. 23)), and the proportion of solid asphalt present as a binder in tarmac or aggregate (ASTM D2172 (ref. 29)). The percentage of asphaltene in oil is a basic parameter covered under the umbrella of SARA analysis (saturates, aromatics, resin, asphaltene). In this approach asphaltene is effectively defined by the method used to recover it from petroleum; asphaltene is obtained by diluting oil with an n-alkane solvent until the asphaltene is no longer solvated (typically a 40-fold dilution) and precipitates from solution.23 Overall, while there is a reasonable correlation (r = 0.90 for n = 11) between SERS- and SARA-calculated percentages of asphaltene in bitumen extracted from the mudrock there is considerable scatter (Fig. 5a); it would not be practical to distinguish 45 from 30% percentage asphaltene for example, and higher values appear systematically over-predicted. It would be practical to distinguish a 50% from a 20% asphaltene concentration. The mass of asphaltene per g of sediment better correlates (r = 0.98 for n = 11) – likely reflecting that the error introduced by weighing the volatile fractions of petroleum (the saturate, aromatic and resin fraction) does not apply to weighing inorganic sediment which is not volatile. In a few instances the amount of asphaltene has been overestimated (Fig. 5b). Of the two assay-types and methods of reporting, given that the bitumen is attached to sediment or rocks and not present as a liquid or free flowing oil phase, reporting the mass of asphaltic petroleum per gram of sediment is more appropriate, and in this instance also the more reliable measure.3.4 Quantification within a natural science context
The concentration of diasterane (a resistant petroleum biomarker2) per gram of sediment is lowest in the sediment-samples that also have the lowest concentration of asphaltene (Fig. 6). Part of the difference is caused by dilution; for example shell fragments and rock types other than petroleum-bearing formations are present in the beach sand and some samples plot close to a mixing line calculated for mixtures of petroleum-bearing formation and petroleum-free inorganic sediment. However, many samples contain far less biomarker than can be explained by dilution, an affect that is even more pronounced for regular steranes (steranes are not as chemically resistant to weathering at surface conditions as diasteranes2). In this case sterane concentrations are tens of thousands of times lower whereas asphaltene concentrations are a hundreds times lesser in the most weathered samples. Therefore, the SERS assay has detected the effects of weathering of asphaltic petroleum within sediments, an effect that can also be seen for other petroleum components such as sterane biomarkers measured by conventional methods. | ||
| Fig. 6 Graphical comparison between standard and SERS-based methods of analysing asphaltene and asphalt. (a) Comparison between ASTM D2414 and SERS based assay, and (b) between a total extraction-based quantification of bitumen content (ASTM D2172/D2172M-17e1) and SERS quantification of bitumen per gram of sediment. Dashed lines show 10% margins. | ||
To provide further context for what is a meaningful difference in concentration, shown in Fig. 6 are end-members for asphalt–sediment mixtures; a sample of deep marine mud with a small amount of both petroleum biomarker and asphaltene and a sample of wearing course asphalt (surface layer asphalt) that has a high amount of asphaltene and low amount of petroleum biomarker. Deep water sediments are the most distal destination for any naturally eroded petroleum-baring sediment, with relatively little terrestrial sediment being transported to the abyssal plane, and such sediment experiencing a long transit,30 as well as mixing with other sediment types. It is notable that deep marine mud plots in a region that by visual extrapolation of the series might be inferred to be genetically related to the Brough sediment (e.g. it would lie at the end of data series).
The wearing course asphalt lies in a region of the graph distinct from both pristine samples of pristine petroleum-baring rocks and petroleum-bearing sediment weathered from the rocks. The asphalt is a human-made material formed by isolating the asphaltic fractions of petroleum from other components, and mixing the isolate with aggregate (crushed rock, analogous to sediment). Such a process may seem highly artificial, but it is not without natural precedence and a naturally occurring example would be tar sand deposits from Alberta, Canada. Tar sands form by cycles of biodegradation and migration of petroleum in the near subsurface;31 during these cycles biodegradation selectively enriches petroleum in asphaltic components by removing lighter components (including terpenoid biomarkers), prior to influxes of new petroleum that contain both biomarkers and asphaltene. Samples of Canadian tar sand, held to have formed by such a process, plot between the highly asphaltic wearing coarse and samples of petroleum reservoir and source rock (Fig. 7), consistent with their formation by enrichment in asphaltic petroleum and loss of volatile biomarkers.
 | ||
| Fig. 7 Concentrations of diasteranes, steranes and asphaltic petroleum in sediment vs. concentration of asphaltene in sediments. | ||
3.5 Discussion
Prior methods for deconvolving Raman spectra of sedimentary organic matter have generally sought to fit multiple individual curves to composite Raman bands; (e.g. the D1–D3 & G bands32,33). This leaves degrees of freedom with respect to the number of curves, their shape and position, and permits deconvolution of complex mixtures. Within the context of surface enhanced Raman scattering deconvolution has previously been done for mixtures of Raman-active pigments within endoliths;11 determining the relative contributions of ββ-carotene, scytonemin and other carotenoids to SERS-spectra measured on solvent extracts from rocks containing pigmented micro-organisms. Taking a composite band at 1176 cm−1 as an example; curves were initially fitted for the ν1 band of carotenoids (1156 cm−1) and for a strong scytonemin band at (1172 cm−1). After this several other curves were added to explain the composite band to represent contributions from the degradation-products of carotenoids,7 structurally similar homologous carotenoids,34 and isomers of scytonemin formed by heating during sample processing. There is also the possibility that bands have asymmetrical shapes naturally or acquire them due to analytical artefacts (i.e. surface induced characteristics caused by chemical interaction with the substrate). So, it can be argued, that the list of “uncharacterised”4 interfering compounds is potentially very large indeed. Therefore, the use of deconvolution procedures that have high degrees of freedom can be justified for sedimentary organic matter because of the range of compounds that may be present.However, for any deconvolution approach in which there is a such a high degree of freedom overfitting remains a genuine and persistent problem, albeit one that can be identified and for which there are solutions. These include both methodological34,35 and the utilisation of common approaches be they algorithmically or systematically applied and enforced,36,37e.g. sharing reference materials within a user group. The approach taken here (using a minimal number of entire surface enhanced Raman spectra) greatly reduces the risk of overfitting because of the limited degrees of freedom involved, and this makes the method of specific use to the automated screening of high numbers of sediment-samples. Without overfitting reasonable (albeit certainly not superior) fits to standard methods can be obtained for sediments containing asphaltic petroleum by removing the contributions from humic and pigmented sedimentary organic matter. This suggests that while it may be justified to account for a wide range of interfering compounds in surface enhanced Raman spectra11 of sediments, the improvement is not necessary for functional data and that the risk of overfitting need not be incurred.
A case can therefore be made, that fully explaining all variance in surface enhanced Raman spectra by small and arbitrary adjustments to curves fitted to explain Raman bands is not necessary to quantify asphaltic petroleum. However, such variance is still present in data, and ultimately if such variance is not going to be explained it will manifest as a source of error that limits reproducibility. This is illustrated in Fig. 5b, where similar detector responses are observed for quantities of asphaltene from 1 to 3 μg. Hyphenation, the combining a SERS detector with a separation stage,14 has been proposed as a method to improve SERS and SERRS analyses of sediments because it permits the isolation of analytes from an interfering sample matrix – e.g. removing the influence of all carotenoids, not just those identified a priori. If successfully applied hyphenated methods could reduce variance in spectral-features due to small amounts of interfering compounds (e.g. the scatter seen in Fig. 4b), which in-turn might improve reproducibility (Fig. 6). Better separation and chemical processing are not the only option and it is likely that better detector components, in this case SERS substrates, could also improve reproducibility,24 to the point where a standard analysis method could be proposed.
4 Conclusions
The asphaltic components of petroleum can be quantified within sediments by using look-up tables to deconvolve surface enhanced Raman spectra. For the case of petroleum-baring rocks being eroded at a beach in Northern Scotland it can be shown that main SERS-active components aside from the asphaltene within the petroleum are humic compounds and pigments such as phycocyanin and carotenoids. Once these components are accounted for the quantification is suitably reliable that it has reasonable correlation with concentrations determined via ASTM (methods), and the data can be used to support comparative natural science data acquired via alternative methods such as GC-MS.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work used samples and data collected during International Ocean Discovery Program (IODP) Expedition 368. SAB gratefully acknowledges NERC award NE/R002576/1; Measuring Rates of Weathered Petroleum Accumulation, South China Sea.Notes and references
- Z. Wang and M. F. Fingas, Development of oil hydrocarbon fingerprinting and identification techniques, Mar. Pollut. Bull., 2003, 47, 423–452 CrossRef CAS PubMed.
- K. E. Peters, C. C. Walters and J. M. Moldowan, The biomarker guide, Cambridge University Press, Cambridge, UK, 2005, vol. 2 Search PubMed.
- J. Connan, Biodegradation of crude oils in reservoirs, Advances in Petroleum Geochemistry, ed. J. Brooks and D. Welte, Academic Press, London, 1984, vol. 1, pp. 299–335 Search PubMed.
- J. I. Hedges, G. Eglinton, P. G. Hatcher, D. L. Kirchman, C. Arnosti, S. Derenne, R. P. Evershed, I. Kögel-Knabner, J. W. De Leeuw, R. Littke, W. Michaelis and J. Rullkötter, The molecularly-uncharacterized component of nonliving organic matter in natural environments, Org. Geochem., 2000, 31(10), 945–958 CrossRef CAS.
- J. G. Speight, The Chemistry and Technology of Petroleum, Taylor and Francis Group, Florida, 5th edn, 2014 Search PubMed.
- D. Zhan and J. B. Fenn, Int. J. Mass Spectrom., 2000, 194, 197–208 CrossRef CAS.
- D. J. Repeta and R. B. Gagosian, Carotenoid diagenesis in recent marine sediments-I. The Peru continental shelf (15°S, 75°W)* * Woods Hole Oceanographic Institution Contribution No. 6361, Geochim. Cosmochim. Acta, 1987, 51(4), 1001–1009 CrossRef CAS.
- J. S. Sinninghe Damsté and M. P. Koopmans, The fate of carotenoids in sediments: An overview, Pure Appl. Chem., 1997, 69(10), 2067–2074 Search PubMed.
- M. Bjorøy, P. B. Hall, R. Løberg, J. A. McDermott and N. Mills, Hydrocarbons from non-marine source rocks, Org. Geochem., 1988, 13(1–3), 221–244 CrossRef.
- J. M. Fulton, M. A. Arthur and K. H. Freeman, Subboreal aridity and scytonemin in the Holocene Black Sea, Org. Geochem., 2012, 49, 47–55 CrossRef CAS.
- R. Wilson, P. Monaghan, S. A. Bowden, J. Parnell and J. M. Cooper, Surface-Enhanced Raman Signatures of Pigmentation of Cyanobacteria from within Geological Samples in a Spectroscopic-Microfluidic Flow Cell, Anal. Chem., 2007, 79, 7036–7041 CrossRef CAS PubMed.
- E. Vogel, R. Geßner, M. H. B. Hayes and W. Kiefer, Characterisation of humic acid by means of SERS, J. Mol. Struct., 1999, 482–483, 195–199 CrossRef CAS.
- O. O. Alabi, A. N. F. Edilbi, C. Brolly, D. Muirhead, J. Parnell, R. Stacey and S. A. Bowden, Asphaltene detection using surface enhanced Raman scattering (SERS), Chem. Commun., 2015, 51(33), 7152–7155 RSC.
- S. A. Bowden, R. Wilson, J. M. Cooper and J. Parnell, The use of surface-enhanced raman scattering for detecting molecular evidence of life in rocks, sediments, and sedimentary deposits, Astrobiology, 2010, 10(6), 629–641 CrossRef CAS PubMed.
- H. Schmidt, N. Bich Ha, J. Pfannkuche, H. Amann, H.-D. Kronfeldtand and G. Kowalewska, Mar. Pollut. Bull., 2004, 49, 229–234 CrossRef CAS PubMed.
- S. A. Bowden, R. Wilson, C. Taylor, J. M. Cooper and J. Parnell, The extraction of intracrystalline biomarkers and other organic compounds from sulphate minerals using a microfluidic format – a feasibility study for remote fossil-life detection using a microfluidic H-cell, Int. J. Astrobiol., 2007, 6(1), 27–36 CrossRef CAS.
- J. Pfannkuche, L. Lubecki, H. Schmidt, G. Kowalewska and H.-D. Kronfeldt, The use of surface-enhanced Raman scattering (SERS) for detection of PAHs in the Gulf of Gdańsk (Baltic Sea), Mar. Pollut. Bull., 2012, 64, 614–626 CrossRef CAS PubMed.
- G. Demaison and B. J. Huizinga, Genetic classification of petroleum systems, United States, N. P., 1991 Search PubMed.
- M. Baba, J. Parnell, D. Muirhead and S. Bowden, Oil charge and biodegradation history in an exhumed fractured reservoir, Devonian, UK, Mar. Pet. Geol., 2019, 101, 281–289 CrossRef CAS.
- Z. Sun, Z. Jian, J. M. Stock, H. C. Larsen, A. Klaus, C. A. Alvarez Zarikian and The Expedition 367/368 Scientists, Proceedings of the International Ocean Discovery Program, South China Sea Rifted Margin, College Station, TX (International Ocean Discovery Program), 2018, vol. 367/368, DOI:10.14379/iodp.proc.367368.2018.
- S. A. Bowden, University of Newcastle upon Tyne and School of Civil Engineering Geosciences, The Molecular Characterisation of Sedimentary Organic Matter and Petroleum by Catalytic Hydropyrolysis, 2004, Print Search PubMed.
- ASTM D3279, Standard Test Method for n-Heptane Insolubles, ASTM International, West Conshohocken, PA, 2012, http://www.astm.org Search PubMed.
- ASTM D4124, Standard Test Method for Separation of Asphalt into Four Fractions, American Society for Testing and Materials, West Conshohocken, PA, 2000, http://www.astm.org Search PubMed.
- X.-M. Lin, Y. Cui, Y.-H. Xu, B. Ren and Z.-Q. Tian, Surface-enhanced Raman spectroscopy: Substrate-related issues, Anal. Bioanal. Chem., 2009, 394, 1729–1745 CrossRef CAS PubMed.
- F. Tuinstra and J. L. Koenig, Raman spectrum of graphite, J. Chem. Phys., 1970, 53, 1126–1130 CrossRef CAS.
- J. D. Pasteris and B. Wopenka, Necessary, but Not Sufficient: Raman Identification of Disordered Carbon as a Signature of Ancient Life, Astrobiology, 2003, 3, 727–738 CrossRef CAS PubMed.
- D. D. Wynn-Williams and H. G. M. Edwards, Proximal Analysis of Regolith Habitats and Protective Biomolecules in Situ by Laser Raman Spectroscopy: Overview of Terrestrial Antarctic Habitats and Mars Analogs, Icarus, 2000, 144, 486–503 CrossRef CAS.
- K. Schiedt and S. Liaaen-Jensen, Chapter 5: Isolation and Analysis, pp 81-108, in Carotenoids Volume 1 A: Isolation and Analysis, ed. G. Britton, S. Liaaen-Jensen and H. Pfander, Pub BirJauser Verlag, Switzerland, 1995 Search PubMed.
- ASTM D2172 and D2172M-17e1, Standard Test Methods for Quantitative Extraction of Asphalt Binder from Asphalt Mixtures, ASTM International, West Conshohocken, PA, 2017, http://www.astm.org Search PubMed.
- B. C. Heezen and C. D. Hollister, The face of the deep, Oxford University Press, New York, 1972 Search PubMed.
- S. Larter, J. Adams, J. D. Gates, B. Bennett and H. Huang, J. Can. Pet. Technol., 2008, 47(1), 52–61 CAS.
- O. Beyssac, B. Goffé, C. Chopin and J. N. Rouzaud, Raman spectra of carbonaceous material in metasediments: a new geothermometer, J. Metamorph. Geol., 2002, 20, 859–871 CrossRef CAS.
- F. Tuinstra and J. L. Koenig, J. Chem. Phys., 1970, 53, 1126–1130 CrossRef CAS.
- R. Withnall, B. Z. Chowdhry, J. Silver, H. G. M. Edwards and L. F. C. de Oliveira, Raman spectra of carotenoids in natural products, Spectrochim. Acta, Part A, 2003, 59, 2207–2212 CrossRef.
- K. Noack, B. Eskofier, J. Kiefer, C. Dilk, G. Bilow, M. Schirmer, R. Buchholz and A. Leipertz, Combined shifted-excitation Raman difference spectroscopy and support vector regression for monitoring the algal production of complex polysaccharides, Analyst, 2013, 138(19), 5639–5646 RSC.
- A. J. Smola and B. Schölkopf, Stat. Comput., 2004, 14, 199 CrossRef.
- S. A. Sandford, et al., Organics Captured from Comet 81P/Wild 2 by the Stardust Spacecraft, Science, 2006, 314, 1720 CrossRef CAS PubMed.
- Asphaltenes, Heavy Oils and Petroleomics, ed. O. C. Mullins, et al., Springer, New York, 2007, DOI:10.1007/0-387-68903-6.
- E. Bandurski, Energy Sources, 1982, 6, 47–66 CrossRef CAS.
- H. Abraham, Asphalts and Allied Substances: Their Occurrence, Modes of Production, Uses in the Arts, and Methods of Testing, D. Van Nostrand Co., Inc, New York, 4th edn, 1938 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: S1: example of SERS data and processing script are provided (written for R). See DOI: 10.1039/c9ay01859j |
| ‡ The term asphaltic petroleum has been used in this work to denote both asphaltene precipitated from oil,38 asphaltene precipitated from source rock extracts,39 and asphalt in the sense of petroliferous tars or “bitumens” (bitumen is used to describe tar and used for the solvent extracts of rocks).40 |
| This journal is © The Royal Society of Chemistry 2019 |