
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA microfluidic platform integrating pressure-driven and electroosmotic-driven flow with inline filters for affinity separations†
Weijia
Leng
 ,
Kimberly
Evans
and
Michael G.
Roper
*
,
Kimberly
Evans
and
Michael G.
Roper
*
Department of Chemistry and Biochemistry, Florida State University, 95 Chieftain Way, Dittmer Building, Tallahassee, FL 32306, USA. E-mail: roper@chem.fsu.edu; Tel: +1-850-644-1846
First published on 29th October 2019
Abstract
Pancreatic islets of Langerhans release glucagon to maintain blood glucose levels, and release of this peptide is dysregulated in diabetes mellitus. Although the importance of proper secretion of this peptide has been shown, no measurement of its release at the single islet level has been reported. In previous work, a non-competitive assay for glucagon was developed with a 6 pM limit of detection, low enough to measure from a single islet. To incorporate this method in an online assay, a microfluidic system with several distinct features was developed. To maintain appropriate flow rates in the presence of the high concentration of salt that was required for the assay, a piezo-actuated pressure transducer with inline flow sensors was used to drive sample flow through 80 × 50 μm (width × depth) channels, while electroosmotic flow was used to gate the sample away from a 15 × 5 μm separation channel. Flow rates tested in this system were 50–200 nL min−1 with relative standard deviations (RSDs) ranging from 1–4%. Use of the pressure-driven flow was found to increase the amount of clogs in the system, so a method to incorporate inline filters into the channels was developed. A total of 4 low resistance, inline microfabricated filters were evaluated, with all designs prolonging the operation time of the microfluidic device to more than 4 hours without clogs observed. Use of this system enabled highly reproducible injections (3–6% RSD). During initial incorporation of the non-competitive assay for glucagon, it was determined that Joule heating was problematic and temperature measurements revealed the separation channel increased to more than 50 °C during operation. A 3D-printed manifold was used to hold a Peltier cooler in place on the microfluidic device which produced a 2.6-fold improvement in the amount of the noncovalent glucagon complex that was detected compared to without cooling. These features are expected to be useful for not only long-term monitoring of the glucagon release from islets of Langerhans, but has the potential to be applied to a number of other microfluidic separation-based assays as well.
Introduction
Islets of Langerhans are the endocrine portion of the pancreas, responding to changing glucose levels with the release of glucoregulatory hormones. Glucagon, a 29-amino acid peptide hormone released from α-cells in islets, stimulates hepatic glucose production and plays a critical role in maintaining glucose homeostasis.1 As evidence of the importance that glucagon plays in controlling overall blood glucose levels, suppression of glucagon secretion has been shown to improve glycemic control in both type 1 and type 2 diabetes.2–4 Despite the significant role that glucagon plays in maintaining euglycemia, there is still much to learn about the secretion dynamics and regulatory mechanisms of this hormone from islets of Langerhans.Glucagon is conventionally measured using enzyme-linked immunosorbent assays (ELISA) from batches of islets.5 While these measurements are highly sensitive, measuring release from multiple islets masks the secretion dynamics and may not allow all mechanistic details to be uncovered. Unfortunately, the low amounts of glucagon secreted6,7 make it challenging to measure at the single islet level. Additionally, the peptide itself is known to form aggregates and degrade quickly in aqueous solutions,8 making it difficult to develop an assay.
Microfluidic devices have gained popularity for their ability to monitor secretion from living islets.9–15 In one report,10 glucagon release was measured from 10 islets using a microfluidic-based competitive immunoassay. While this assay had the necessary limit of detection (LOD) and sensitivity for measuring glucagon secretion from these relatively few number of islets, to observe the unconvoluted secretion dynamics of release from a single islet, a lower LOD is required. Recently, we developed a non-competitive assay for measurement of glucagon which had a calculated LOD of 6 pM using a mirror-image DNA/RNA aptamer known as Spiegelmer.‡16 Similar to aptamers or antibodies, Spiegelmers can bind to target molecules such as proteins and peptides with high affinity after a selection process. Spiegelmers use the L-enantiomer of the nucleotide, which makes them more resistant to degradation by nucleases than their D-enantiomeric counterparts.17 Although this LOD is low enough to enable detection of glucagon secretion from a single islet, since the assay was offline, it was not possible to use for monitoring glucagon release dynamics from living islets.
In this manuscript, we describe our initial incorporation of this non-competitive assay into a microfluidic system for eventual use in an online assay. During the development of the assay, a number of problems were encountered, including incompatibility of the sample buffer with the use of electroosmotic flow (EOF) in the microfluidic system, clogging of the device, and the generation of a large amount of Joule heat during the separation that hindered detection of the affinity complex. However, since these problems are not unique to the glucagon assay, but may also interfere with the integration of other analytical assays into microfluidic systems, the solutions that were developed to overcome these problems are expected to be useful to a number of systems.
Experimental
Materials and reagents
Sodium tetraborate and ethylene glycol were obtained from Fisher Scientific (Pittsburgh, PA). Ethylenediaminetetraacetic acid (EDTA), sodium hydroxide (NaOH), hydrochloric acid (HCl), Tween-20, and bovine serum albumin (BSA) were purchased from EMD Chemicals (San Diego, CA). Ethanol was from Koptech (King of Prussia, PA). 4,4-Difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-propionyl ethylenediamine, HCl (Bodipy) was obtained from Setareh Biotech (Eugene, OR). Laser grade rhodamine B (RhB) was from Acros Organics (Pittsburgh, PA). 6-FAM labeled L-DNA/L-RNA anti-glucagon Spiegelmer® (Apt*) (NOX-G16004: 5′-GCGGGOH AAATG GOHGAGOHGOH GCTAG GTOHGGAOH AOHGGAA TCTGA GCGC-3′) was synthesized at NOXXON Pharma (Berlin, Germany).18 Glucagon and all other chemicals were purchased from Sigma-Aldrich (Saint Louis, MO), unless otherwise stated.Sample preparation
All buffers and reagents were prepared using ultrapure deionized (DI) water (NANOpure Diamond™, Barnstead International, Dubuque, IA) and filtered using 0.2 μm nylon syringe filters (Pall Corporation, Port Washington, NY). The separation buffer was 20 mM borate at a pH of 9.0. Bodipy and RhB stock solutions were prepared in DI water, while fluorescein stock solution was prepared in 1 M NaOH. The final concentrations of 1 μM bodipy, 5 μM RhB, and 100 nM fluorescein were made by diluting the stock solutions with separation buffer.Glucagon was reconstituted in degassed DI water with 0.1% (v/v) formic acid. A working solution of glucagon was then made by diluting an appropriate amount of the stock solution into the sample buffer, which consisted of 20 mM tricine, 1 mM EDTA, and 10 mM MgCl2 (pH 7.4) with an additional 0.1% (w/v) BSA and 0.1% (w/v) Tween-20. A 5 μM Apt* stock solution was prepared using 10 mM Tris–HCl, 1 mM EDTA, 1 mM MgCl2, at a pH of 8.0. Prior to mixing with glucagon, a thermal cycler (Bibby Scientific Ltd, Burlington, NJ) was used to heat the Apt* stock solution to 94 °C, held at that temperature for 3 min, then cooled down to room temperature at a rate of 0.1 °C s−1.16 An appropriate volume of the Apt* stock solution was added to the glucagon working solution to give a final concentration of 1 μM and 500 nM of glucagon and Apt*, respectively. The mixture was then incubated in the dark at 4 °C for 48 hours prior to use in the microfluidic device.
Instrumentation
The microfluidic device was fabricated by UV photolithography and wet etching using previously described methods.19 Briefly, the device consisted of both deep (30 μm) and shallow (5 μm) channels that were fabricated on different pieces of glass (Telic, Valencia, CA). Etched channel dimensions were verified using a P-15 Stylus Profilometer (KLA-Tencor, Milpitas, CA). Unless otherwise stated, all dimensions relating to the microfluidic device in the text are the final etched dimensions. Access holes to the channels were drilled on the piece of glass with the deeper channels. The glass pieces were cleaned after removing the remaining photoresist and chrome, aligned under a stereomicroscope, and placed in an oven for thermal annealing at 640 °C for 8 hours. Reservoirs (IDEX Health & Science, Oak Harbor, WA) were then attached to the device according to the manufacturer's instructions.Unless otherwise stated, samples were held in a polypropylene tube and connected to a piezo-actuated pressure controller (OB1, Elveflow, Paris, France). The pressure controller delivered N2 to the top of the sample tube forcing the sample through an 18 cm piece of 100 μm i.d. × 360 μm o.d. capillary (Polymicro Technologies, Phoenix, AZ) to an inline flow sensor (FS, Elveflow, Paris, France), and finally to the inlet of the microfluidic device via a 30 cm length of same dimension capillary. The capillary was connected to the microfluidic device using commercial adapters (IDEX Health & Science, Oak Harbor, WA).
A manifold (Fig. S1†) to hold the microfluidic device and the temperature control elements was printed in-house (Form 2, Formlabs, Somerville, MA). The manifold sandwiched the microfluidic device between a top and a bottom plate that were screwed together. A Peltier cooler (14.8 × 14.5 mm, 8.4 W, TE Technology, Traverse City, MI) was held in the top plate of the manifold and was attached to the microfluidic device using thermal paste (Arctic Silver 5, Visalia, CA). The position of the cooler was fixed by the manifold to start 5 mm after the injection cross and span the majority of the separation channel. A J-type thermal couple (Omega Engineering, Stamford, CT) was attached to the bottom of the microfluidic device under the Peltier cooler. To maintain a constant temperature, a temperature controller (FTC-100D, Accuthermo Technology, Freemont, CA) was used to monitor the temperature from the thermal couple and apply an adjustable voltage to the Peltier. Unless otherwise stated, any temperatures reported in this manuscript were the readings from the thermocouple.
To facilitate continuous operation of the Peltier, an in-house fabricated hollow copper block (1.5 × 1.5 cm) was placed on the Peltier device with thermal paste. Inlet and outlet tubing were attached to the block using barbed fittings and a peristaltic pump was used to cycle ethylene glycol through the block at 1 mL min−1. The tubing that delivered the ethylene glycol to the device was submerged in a beaker of ethanol and ice.
Detection and microfluidic device operation
The assembled manifold with the microfluidic device was placed on the stage of a Nikon TS-100 inverted fluorescence microscope (Nikon Instruments, Melville, NY). All reservoirs besides the sample inlet were loaded with 90 μL of separation buffer, covered by plastic caps, and platinum electrodes inserted. To shunt the sample to waste, +5000 V was applied to the gate reservoir (UltraVolt, South Thief River Falls, MN) while the separation and waste reservoirs were grounded. Injections were performed as described in the text.For detection of separated components, laser-induced fluorescence was performed 2.5 cm downstream from the injection cross. A 488 nm laser line from a 50 mW solid-state laser (Melles Griot, Carlsbad, CA) was used as the excitation source. The light was directed to the microscope via a 6 ft fiber bundle, collimated (CeramOptec, East Longmeadow, NY), and directed to a 538/685 nm bandpass filter (Semrock, Rochester, NY). The light was focused using a 40X, 0.6 NA objective (Nikon Instruments) while the emission was collected by the same objective, passed through the bandpass filter, a spatial filter, and a 550 ± 40 nm bandpass filter (Omega Filters, Brattleboro, VT) before being detected by a photomultiplier tube (PMT). The spatial filter, bandpass filter, and PMT were housed in a photometer (Photon Technology International, Birmingham, NJ). Control of the injections and data collection were performed by a custom LabVIEW Program (National Instruments, Austin, TX).
For measurement of RhB intensity, the output from a Xenon arc lamp (Lambda XL, Sutter Instruments, Novato, CA) was directed to a bandpass filter (535 ± 15 nm, Omega Optical, Brattleboro, VT) and a 565 nm dichroic mirror (Omega Optical) in a Nikon Ti–S inverted microscope. The excitation light was focused onto the microfluidic device using a 10X, 0.5 NA objective (Nikon Instruments). Emission was collected by the same objective, passed through the dichroic mirror, and an emission filter (595 ± 20 nm). Fluorescence images were acquired using a CMOS camera (Qimaging, Surrey, BC, Canada) with a 70 ms exposure every 10 s. Two regions of interest were used to define both the channel and the background. Intensities were calculated by subtracting the value of the background from the channel. The timing of the camera and the excitation shutter, as well as defining the regions of interest, were performed by Nikon NIS Elements software.
To correlate temperature to the RhB intensity,20 all reservoirs and channels in the device were filled with 5 μM RhB. The fluorescence intensity 2.5 cm downstream from the injection cross in the separation channel was measured while the temperature of the Peltier was varied from 17–70 °C using the temperature control system described above. Similar to the previous report,20 the average RhB intensity at each temperature was plotted as a function of applied temperature and fit with a 3rd order equation using Origin 9.0 (Origin Labs, Northampton, MA). To measure the RhB intensity when the separation voltage was applied, the temperature control system was turned off and the sample inlet and gate reservoir were loaded with 90 μL of the RhB sample, while the other two reservoirs were loaded with 90 μL of the separation buffer. The fluorescence intensity 2.5 cm downstream from the injection cross was measured while voltages ranging from 200–5000 V were applied to the gate reservoir and the separation waste reservoir was grounded. Each RhB measurement occurred 3 min after a change in the temperature or voltage. The average RhB intensity at each applied voltage was then converted to temperature using the RhB calibration curve.
Prior to experiments, the microfluidic device was conditioned with 1 M NaOH, DI water, and separation buffer for 15 min each. After experiments, the device was cleaned by flowing DI water through the sample inlet for 20 min, and then stored in DI water.
Data analysis
Electropherograms were analyzed using a previously described method.21 All data are reported as the average ± 1 standard deviation (SD) unless otherwise stated.Results and discussion
During incorporation of the non-competitive assay for glucagon into the microfluidic system, several problems were encountered. Due to the increasing use of microfluidic devices for cellular systems, for example, in microphysiological systems or organ-on-chip applications, we expect that the problems that were encountered would not be unique to this application. Therefore, solutions to these common problems are expected to be of interest to current and future users of these systems.Microfluidic design for implementation of pressure-driven flow
Initial incorporation of the affinity assay for glucagon was performed in a similar manner as our previous experiments for insulin where cellular perfusate and affinity reagents are delivered to a mixing channel via EOF.13,19,22 While EOF provides simplicity in the instrumentation, this delivery method was not successful for the glucagon assay due to a significantly diminished sampling rate. We attributed the low sampling rate to the high concentration (10 mM) of Mg2+ in the sample buffer. The high Mg2+ concentration in the sample buffer is required for stabilizing the structure of Apt*, but divalent cations are known to reduce EOF.23 To circumvent this problem, pressure-driven flow (PDF) was implemented for delivery of the sample to the separation region of the device where EOF was used.Initially, the PDF and EOF channels were made on the same layer of the glass, with all channel widths 20 μm before etching. Different PDF channel lengths from 5–18 cm and etching depths ranging from 5 μm to 20 μm were fabricated and tested. However, in these channels, the pressures required to generate the appropriate flow rates were greater than those that could be produced from the pressure system that was used, so this design was deemed not suitable. In order to couple pressure-driven sample delivery with electrophoretic separation, a mixture of both deep and shallow channels was required.
Similar to previous works that coupled PDF and EOF using flow-through sampling on a microchip,24–26 a larger channel was then tested for the regions on the device where PDF was desired. As is shown in Fig. 1, the perfusion and sample waste channels were 80 × 30 μm (width × depth), while the gate and separation channels, where EOF was used, were and 15 × 5 μm (width × depth). The deep channels were fabricated on separate pieces of glass from the shallow channels. To facilitate alignment of these pieces for thermal annealing, the injection cross was fabricated on the same glass piece as the separation channel, but the length of the cross was such that when the glass piece containing these shallow channels was brought into contact with the piece of glass containing the deep channels, the cross extended into the deep perfusion and sample waste channels by 1000 μm. The overlap of the deep and shallow channels is shown in detail in Fig. 1. This design was found to be critical for facilitating the manual alignment of the different glass pieces during fabrication.
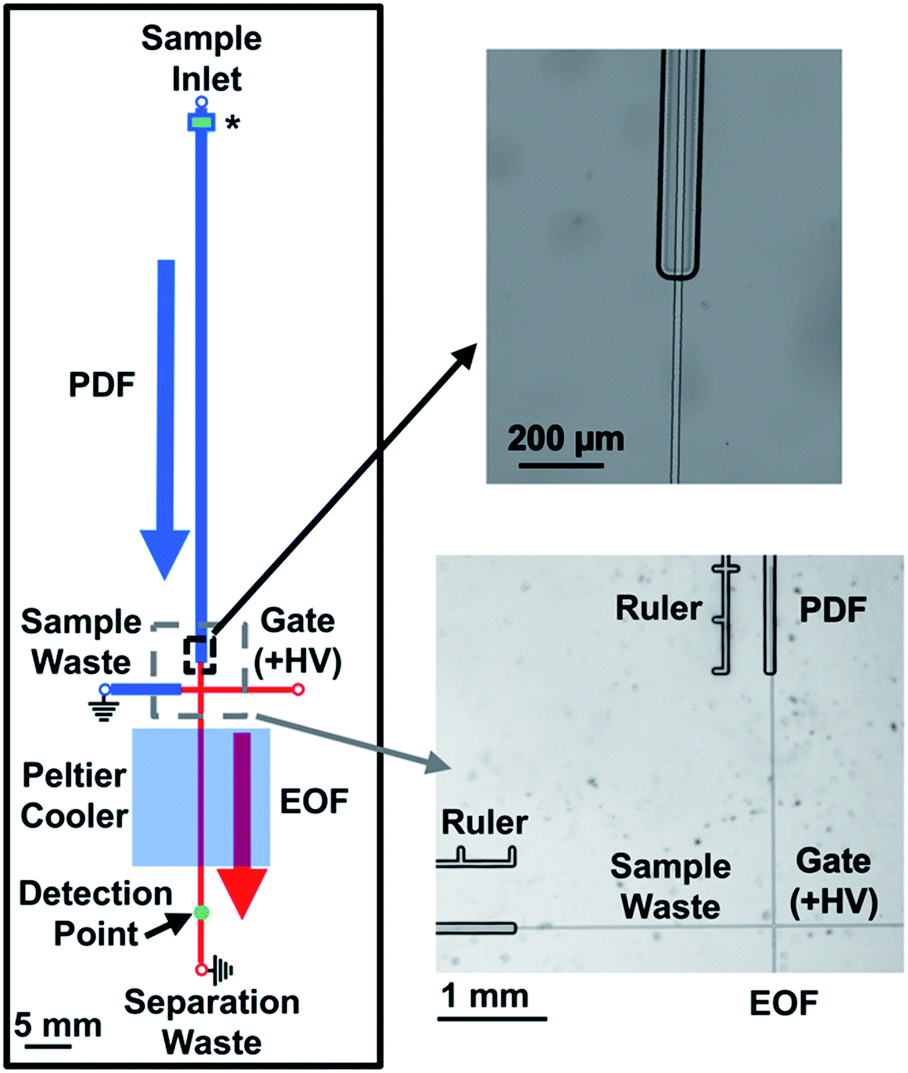 | ||
| Fig. 1 Channel design of the microfluidic device to couple PDF and EOF. All channels shown in blue were fabricated on one piece of glass and were 80 μm in width and 30 μm in depth. All channels shown in red were fabricated on a separate piece of glass and were 15 μm in width and 5 μm in depth. While the lengths of the channels are drawn to scale, the widths of the channels are not; the thicknesses of the lines are meant to only convey the relative size difference between the PDF and EOF channels. PDF was generated by the piezo-actuated pressure regulator (not shown) and delivered to the sample inlet where it flowed through the perfusion channel to the injection cross. EOF was generated by application of +HV to the gate reservoir while the sample and separation waste reservoirs were grounded. At the beginning of the perfusion channel, the sample passed through an inline filter (noted by a (*)) to remove particulates. More details on the filter design are provided in the text and in Fig. 2. The injection cross made with shallow channels extended into the perfusion and sample waste channels by 1000 μm. A zoomed in image of the overlapped intersection is shown on the top right, while an image of the injection cross is shown in the bottom right. The “Ruler” next to the perfusion and sample waste channel was used to help ensure consistent alignment of the two pieces of glass during fabrication. The distances from the end of the perfusion and beginning of sample waste channels to the injection cross were 2500 μm. | ||
To achieve accurate and reproducible PDF rates of the sample, an inline flow sensor was used to feedback to the pressure regulator. Flow rates evaluated with this system were 50–200 nL min−1 and RSDs ranged from 1–4%. To gate the sample away from the separation channel and help direct the PDF to the sample waste channel, +5000 V was applied to the gate reservoir while the remaining reservoirs (excluding the sample reservoir) were grounded. This configuration resulted in EOF from the gate reservoir to the waste and sample waste reservoir. Combined with the difference in the channel sizes of the separation and sample waste channels, even the highest PDF rate of 200 nL min−1 was gated away from the separation channel. To perform an injection, the voltage applied to the gate reservoir was dropped to 200 V for a specified amount of time (the injection time), which enabled the sample to break through the gating flow and load into the separation channel. To re-establish gating flow and commence the separation, the potential on the gate reservoir was increased every 25 ms. The voltage profile increased from 200 to 2000 V in the first 25 ms after completion of the injection time, followed by 1000 V increments over each subsequent 25 ms. By performing this voltage profile, the reproducibility of the injection voltage ramp was improved compared to without a timing sequence which showed voltage spikes and irregularities in the timing.
Incorporation of inline filters
Upon integration of PDF within the device, we began to observe an increase in the number of dust and particulates found in the channels compared to EOF-only devices. These particulates were likely from the environment, such as dust or plastic particles from the reservoir, and not glucagon aggregation since devices became clogged during flushing with even glucagon-free solutions. We hypothesized that it was due to the higher flow rate (50 nL min−1) used with the PDF system compared to what was used in the EOF (∼50 pL min−1) system, since the increased flux of solution would increase the possibility of dust flowing into the channel. The majority of particulates were found at the intersection of the deep and shallow channels, effectively blocking fluid flow into the EOF region.A previous report used microfabricated filters to prevent dust and other particles from entering a microfluidic channel.27 Similarly, to reduce clogs in our microfluidic device, we incorporated inline microfabricated features to act as filters. These filters consisted of an array of pillars with different sizes, the pattern of which are described in more detail below. To ensure that they removed particles that would clog the EOF channels, the minimum dimension between the pillars was set to 15 μm, i.e. the width of the EOF channels. Since conventional HF etching was used to fabricate the device, the pillar array could not be placed in the large perfusion channels as that would require high aspect ratio fabrication techniques. Instead, the filters were etched on the same glass piece as the shallow EOF channels, while the PDF channel that delivered the sample to the injection cross was fabricated with an ∼700 μm unetched region. Similar to the alignment of the shallow injection cross with the deep perfusion and sample waste channels, the deep PDF channels slightly overlapped the shallow filter region (Fig. 2A). This configuration ensured the sample went through the shallow filter region removing any particulates that may clog the EOF channels, and then back into the deep channels.
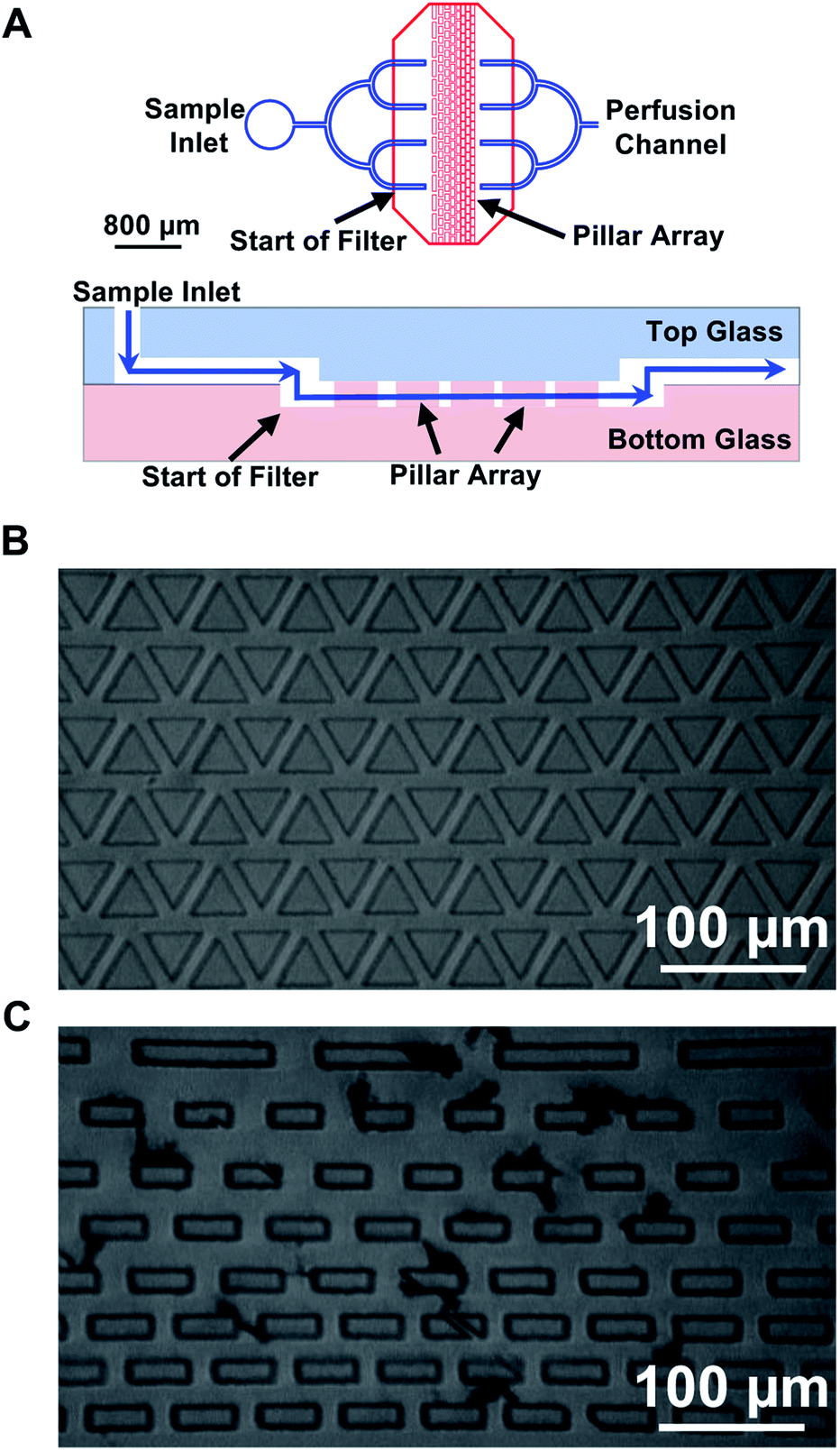 | ||
| Fig. 2 Image of inline filters. (A) As shown in the top image, within 500 μm of the sample inlet, the perfusion channel (blue), fabricated on the top piece of glass, split into four branches. The ends of these branches overlapped 400 μm past the filter region which was fabricated on the bottom piece of glass with the filter features (red). The output of the filter was the opposite of the input with four branches merging back into the single perfusion channel. A side-view (not to scale) of the overlapped perfusion channel with the filter region is shown in the bottom image. The blue line shows the liquid path traveling in the perfusion channel on the top piece of glass, into the bottom piece to be filtered, and then back to the top piece. (B) A filter with triangular pillars and an equal spacing pattern is shown. (C) An image of rectangular pillars with a decreasing spacing pattern was taken after operating for 2 weeks. Particulates had accumulated between the pillar arrays, but did not hinder the PDF. | ||
A total of four different microfabricated inline filters were evaluated, all containing pillars with different shapes and spacing. The filters were categorized into two types based on the shape of the pillar: triangular (Fig. 2B) and rectangular (Fig. 2C). The triangular filters consisted of 6 rows of equilateral triangles with a side length of 100 μm, the rectangular filters were made of 8 rows of rectangles with a width of 50 μm, the first row having rectangles with a length of 200 μm, and the next two rows with a length of 90 μm and the last 5 rows having a length of 100 μm. In each category, the pillars had two spacing patterns. The first pattern had equal spacing (15 μm) between each feature (Fig. 2B), the second pattern had decreased spacing between the features, from 70 μm in the first row to 15 μm in the last three rows (Fig. 2C).
During delivery of 100 nL min−1 flow through the device, the pressure was ∼20% higher (total pressure of 5–6 psi) compared to channels without filters. There was essentially no difference in pressure between the various filter types. Even though the PDF was driven through the shallow filter area, this distance was short, and furthermore, the flow paths acted as many resistors in parallel, reducing the resistance of the entire filter. This contributed to the relatively small increase in pressure observed in the presence of the filter.
All designs fabricated were successful at reducing the number of clogs that occurred in the system, even after more than 4 hours of continuous operation of the device. Several devices were operated for more than 2 weeks, and although there was an accumulation of some particles in the filter region (Fig. 2C), the devices could continue to be used. This was a substantial improvement in the lifetime of the devices, as before the filters were implemented, the devices would clog within ∼1 hour of use. When necessary, the accumulated particles could be removed by applying PDF through the sample waste channel, which pushed the particles out via the sample inlet. A caveat to the use of the filters was when first using a new device, air bubbles could be introduced to the filter region, but these were easily removed through flushing with DI water using PDF. While no difference was observed between the filter designs, we chose to use the design shown in Fig. 2C for the remainder of the experiments.
Once the coupling of the PDF and EOF system was achieved and the filters were in place, we set out to examine the reproducibility of the new injection method. In order to determine the injection reproducibility, pressure injections were performed using a premixed solution of 1 μM bodipy and 100 nM fluorescein. The sample was delivered to the injection cross at a rate of 100 nL min−1 and an injection time of 1.8 s was used.
The peak heights of bodipy and fluorescein for 60 consecutive runs were recorded and the ratio of the heights are shown in Fig. 3. An example electropherogram of the separation is shown in Fig. S2.† The different colors show the results from three independent experiments that were performed on one microfluidic device. In each experiment, new separation buffers and samples solutions were used, requiring disconnection and reconnection of the sample tubing to the microfluidic device, therefore the multiple experiments demonstrate the reproducibility of the system. The peak height ratio of the two compounds were consistent within and between experiments with the relative standard deviation for the three experiments ranging from 3.3–6.3% (Table S1†), indicating that the PDF injection system produced stable and reproducible injections.
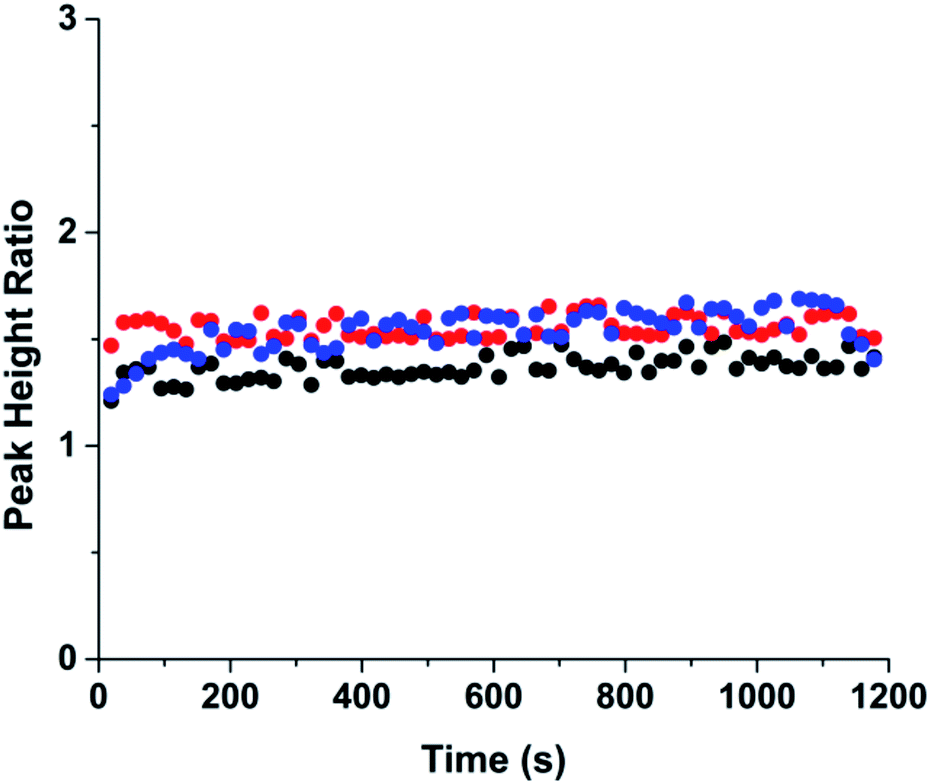 | ||
| Fig. 3 Injection reproducibility. The peak height ratio of bodipy and fluorescein are shown across 60 injections. Three sets of experiments were performed with each color representing one set of experiment. Injections were performed every 20 s for using a PDF rate of 100 nL min−1, an injection time of 1.8 s, and a separation voltage of 5000 V. | ||
Incorporation of glucagon assay
Following these characterizations, the glucagon assay was then tested with the microfluidic device. The assay was similar to that described previously16 and used Apt* as the binding agent to glucagon, producing the bound Apt*-glucagon and free Apt* peak. In the initial incorporation of the method using the combined PDF and EOF system described above, a small amount of the bound Apt*-glucagon complex was observed compared to the amount of free Apt*. Because the amount of bound Apt* at a fixed concentration of glucagon is inversely proportional to the LOD of the assay, increasing the amount of bound Apt* is expected to decrease the LOD and thereby improve the assay. Because increased separation temperatures from Joule heating have been shown to be detrimental to the bound affinity complex,28–30 we set out to determine the temperature of the separation during normal operation, and implement a cooling system if necessary.To determine if there was an increase in the temperature of the separation channel during operation, the temperature during the separation channel under normal operating conditions needed to be measured. The relationship between the separation voltage and temperature was quantified using RhB, a fluorescent dye whose quantum yield is dependent on temperature.20,31 An initial calibration curve of RhB and temperature was first obtained using a Peltier to heat the microfluidic device to known temperatures while the RhB fluorescence was measured in the separation channel. The calibration curve of RhB with different temperatures is shown in Fig. 4A. Each point is the average intensity from a 5 minute measurement with error bars corresponding to ±1 SD. Fig. S3† shows the overall image intensity for each of the temperature in Fig. 4A. As temperature increased, the measured intensity of RhB decreased, similar to what others have reported.20,32,33
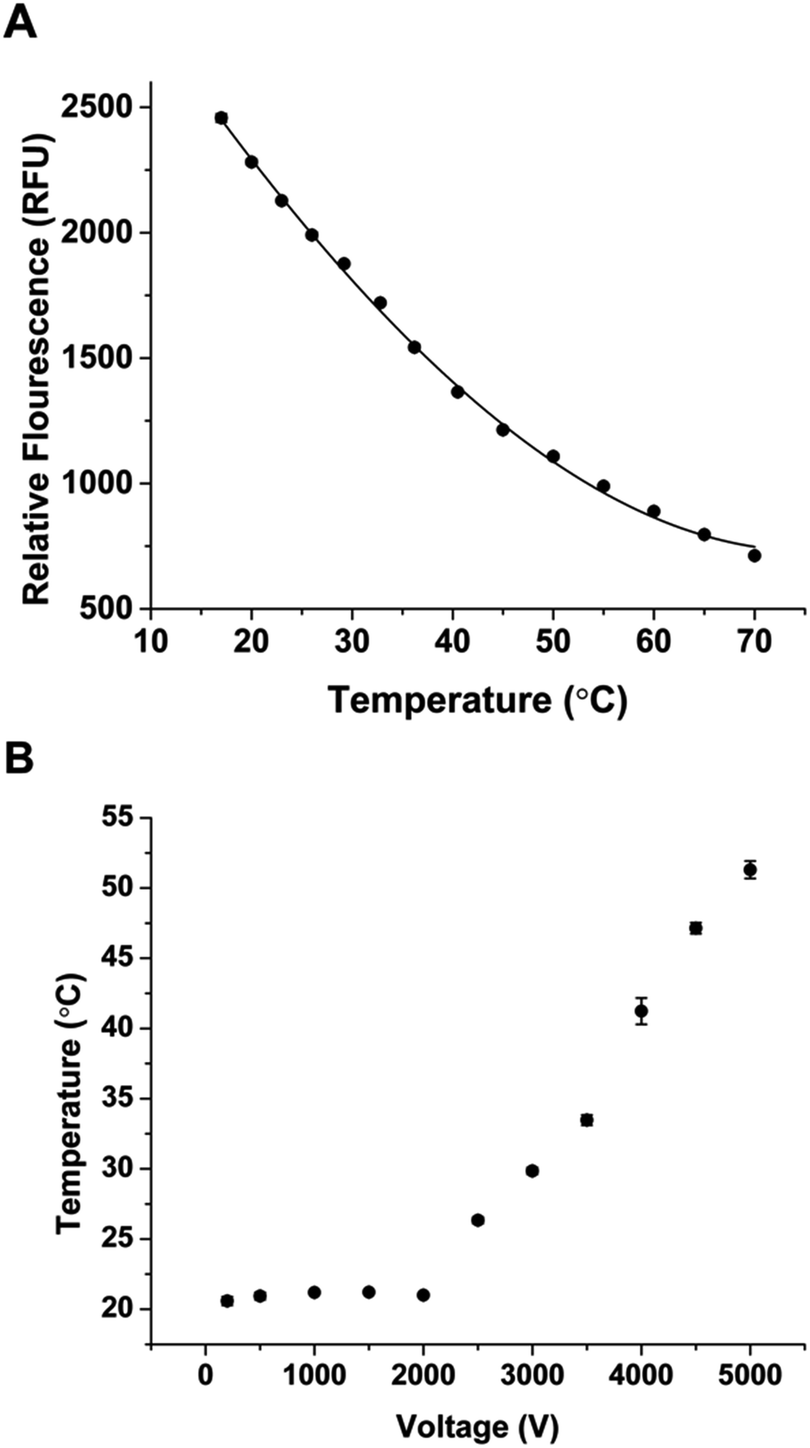 | ||
| Fig. 4 Temperature measurements using RhB. (A) The RhB fluorescence was measured as a function of temperature in the microfluidic device, with each data point representing the average intensity of 30 images and error bars representing ±1 SD. The solid line is a best fit curve to the data using a third order polynomial with an equation: RFU = 0.00156T3 + 0.246T2 − 63.5T + 3451. (B) The RhB intensity in the microfluidic device was measured as a function of applied separation voltage and was converted to temperature using the calibration in (A). | ||
Once the calibration curve was obtained, the Peltier was turned off and the separation voltage was applied while monitoring the intensity of RhB. Fig. 4B shows the measured temperature at different separation voltages. When the voltage was below 2000 V, the temperature was stable at ∼21 °C indicating that the heat generated at these voltages was able to be dissipated. When the voltage was increased above 2000 V, heating increased in a linear fashion up to the maximum voltage tested. At 5000 V, the measured temperature inside the separation channel was more than 50 °C. These elevated temperatures are similar to what has been reported during electrophoretic separations.20 Because of these elevated temperatures, we expected that a temperature control system would increase the amount of bound Apt*-glucagon by preventing its dissociation during the separation.
Initial integration of the temperature system was achieved using a Peltier cooler over the separation channel. Long-term operation of the Peltier was not possible due to excessive heat that built up on the opposite side. The best approach to remove this excessive heat was to place a hollow copper block on top of the Peltier and circulate coolant through the block (Fig. S1†). The improved temperature control system was able to reduce the temperature to as low as 15 °C. However, long-term operation at such a low temperature resulted in condensation forming on the copper block, which was deemed too dangerous to operate because of the potential for electrical discharge. Instead, the temperature was set to 21 °C and the temperature control system was able to consistently keep the measured temperature to ±0.1 °C for at least one hour. The separation was then attempted at 21 °C, and as shown in Fig. 5, produced an increased ratio of bound Apt*-glucagon to free Apt* compared to the separation performed without active temperature control. A slower migration velocity can also be seen in the cooled separation compared to the uncooled separation. This longer time in the electric field also resulted in an improvement in the resolution of the peaks, which should help in quantitation.
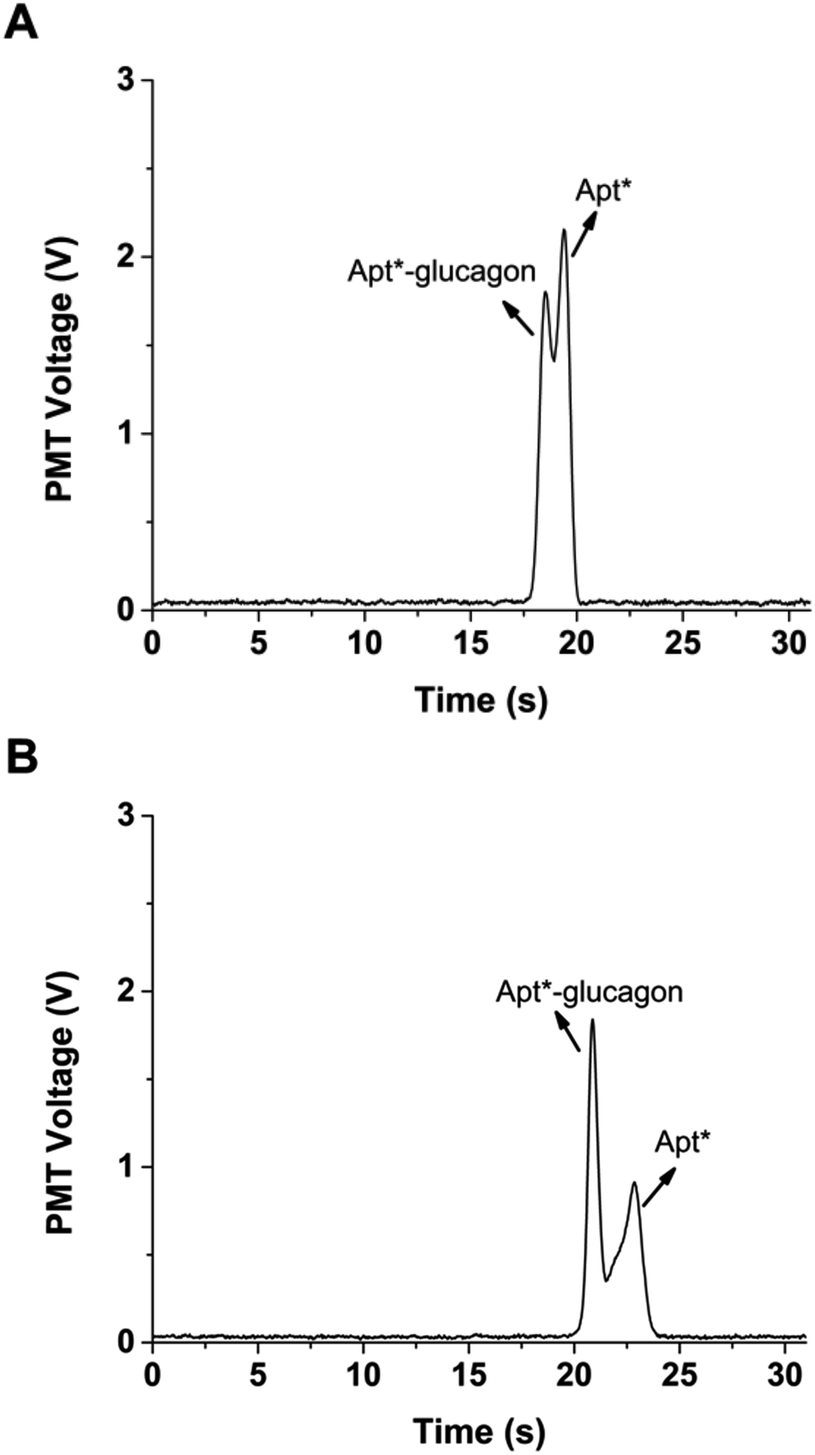 | ||
| Fig. 5 Glucagon non-competitive assay. (A) Representative electropherogram of Apt*-glucagon and Apt* separation without cooling. The bound Apt*-glucagon and free Apt* peaks are labeled. Across 15 runs, the average (±SD) ratio of the two peaks was 0.84 ± 0.01. The PDF rate was 50 nL min−1 and a 1 s injection time was used, field strength for separation was 1250 V cm−1. (B) A representative electropherogram using the same injection conditions as in (A), but with a separation temperature of 21 °C. Across 15 runs, the average (±SD) ratio of the bound Apt*-glucagon to free Apt* peaks was 2.22 ± 0.05, a significant increase compared to the separations performed without temperature control (p < 0.01, 1-tailed Student t-test). | ||
Conclusions
This work demonstrates several features that were used to facilitate a non-competitive assay for glucagon into a microfluidic system. We expect that these features will be useful to other groups who are attempting to develop affinity assays to enable monitoring of cellular secretions. The use of PDF for delivery of the high salt solution was essential and opens the possibility for using other buffers for optimization of binding conditions. Moreover, the use of PDF enabled reproducible sample introduction. The caveat to the PDF was the observation of clogging, but was mitigated by inline filters that allowed continuous operation of the microfluidic device for several hours. Finally, the use of high voltage for the separation produced temperatures that may be incompatible with stabilization of the non-covalent affinity complexes, but this effect can be lowered using active temperature control systems. The described microfluidic system improved the separation of the glucagon assay, however, in order to monitor glucagon secretion from a single islet, the assay itself, such as incubation conditions, needs continued improvement. In the future, with the optimized glucagon assay, it could be used for quantifying glucagon release from islets of Langerhans to elucidate their secretion dynamics under various stimuli.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank NOXXON Pharma for donation of the Spiegelmer®. This work was supported in part by grants from the National Institutes of Health (R01 DK080714) and using resources and/or funding provided by the NIDDK-supported Human Islet Research Network (HIRN, RRID:SCR_014393; https://hirnetwork.org; UC4 DK116283).Notes and references
- R. P. Robertson, Diabetes, 2010, 59, 2735–2736 CrossRef CAS PubMed.
- R. H. Unger and L. Orci, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 16009–16012 CrossRef CAS PubMed.
- J. E. Campbell and D. J. Drucker, Nat. Rev. Endocrinol., 2015, 11, 329–338 CrossRef CAS PubMed.
- D. D'alessio, Diabetes, Obes. Metab., 2011, 13, 126–132 CrossRef PubMed.
- T. Katsura, D. Kawamori, E. Aida, T.-A. Matsuoka and I. Shimomura, PLoS One, 2017, 12, e0176271 CrossRef PubMed.
- A. Salehi, C. D. de la Cour, R. Håkanson and I. Lundquist, Regul. Pept., 2004, 118, 143–150 CrossRef CAS PubMed.
- C. Guillo and M. G. Roper, J. Chromatogr. A, 2011, 1218, 4059–4064 CrossRef CAS PubMed.
- N. Caputo, J. R. Castle, C. P. Bergstrom, J. M. Carroll, P. A. Bakhtiani, M. A. Jackson, C. T. Roberts Jr, L. L. David and W. K. Ward, Peptides, 2013, 45, 40–47 CrossRef CAS PubMed.
- M. G. Roper, J. G. Shackman, G. M. Dahlgren and R. T. Kennedy, Anal. Chem., 2003, 75, 4711–4717 CrossRef CAS PubMed.
- J. G. Shackman, K. R. Reid, C. E. Dugan and R. T. Kennedy, Anal. Bioanal. Chem., 2012, 402, 2797–2803 CrossRef CAS PubMed.
- A. M. Clark, K. M. Sousa, C. N. Chisolm, O. A. MacDougald and R. T. Kennedy, Anal. Bioanal. Chem., 2010, 397, 2939–2947 CrossRef CAS PubMed.
- C. J. Easley, J. V. Rocheleau, W. S. Head and D. W. Piston, Anal. Chem., 2009, 81, 9086–9095 CrossRef CAS PubMed.
- B. Bandak, L. Yi and M. G. Roper, Lab Chip, 2018, 18, 2873–2882 RSC.
- J. F. Dishinger, K. R. Reid and R. T. Kennedy, Anal. Chem., 2009, 81, 3119–3127 CrossRef CAS PubMed.
- A. R. Lomasney, L. Yi and M. G. Roper, Anal. Chem., 2013, 85, 7919–7925 CrossRef CAS PubMed.
- L. Yi, X. Wang, L. Bethge, S. Klussmann and M. G. Roper, Analyst, 2016, 141, 1939–1946 RSC.
- A. Vater and S. Klussmann, Drug Discovery Today, 2015, 20, 147–155 CrossRef CAS PubMed.
- A. Vater, S. Sell, P. Kaczmarek, C. Maasch, K. Buchner, E. Pruszynska-Oszmalek, P. Kolodziejski, W. G. Purschke, K. W. Nowak, M. Z. Strowski and S. Klussmann, J. Biol. Chem., 2013, 288, 21136–21147 CrossRef CAS PubMed.
- L. Yi, X. Wang, R. Dhumpa, A. M. Schrell, N. Mukhitov and M. G. Roper, Lab Chip, 2015, 15, 823–832 RSC.
- D. Ross, M. Gaitan and L. E. Locascio, Anal. Chem., 2001, 73, 4117–4123 CrossRef CAS PubMed.
- J. G. Shackman, C. J. Watson and R. T. Kennedy, J. Chromatogr. A, 2004, 1040, 273–282 CrossRef CAS PubMed.
- L. Yi, B. Bandak, X. Wang, R. Bertram and M. G. Roper, Anal. Chem., 2016, 88, 10368–10373 CrossRef CAS PubMed.
- S. Datta, A. T. Conlisk, H. F. Li and M. Yoda, Mech. Res. Commun., 2009, 36, 65–74 CrossRef CAS PubMed.
- S. Attiya, A. B. Jemere, T. Tang, G. Fitzpatrick, K. Seiler, N. Chiem and D. J. Harrison, Electrophoresis, 2001, 22, 318–327 CrossRef CAS PubMed.
- Y.-H. Lin, G.-B. Lee, C.-W. Li, G.-R. Huang and S.-H. Chen, J. Chromatogr. A, 2001, 937, 115–125 CrossRef CAS PubMed.
- S.-H. Chen, Y.-H. Lin, L.-Y. Wang, C.-C. Lin and G.-B. Lee, Anal. Chem., 2002, 74, 5146–5153 CrossRef CAS PubMed.
- B. Wunderlich, D. Nettels, S. Benke, J. Clark, S. Weidner, H. Hofmann, S. H. Pfeil and B. Schuler, Nat. Protoc., 2013, 8, 1459–1474 CrossRef CAS PubMed.
- M. U. Musheev, Y. Filiptsev, V. Okhonin and S. N. Krylov, J. Am. Chem. Soc., 2010, 132, 13639–13641 CrossRef CAS PubMed.
- D. D. Buchanan, E. E. Jameson, J. Perlette, A. Malik and R. T. Kennedy, Electrophoresis, 2003, 24, 1375–1382 CrossRef CAS PubMed.
- N. Mukhitov, L. Yi, A. M. Schrell and M. G. Roper, J. Chromatogr. A, 2014, 1367, 154–160 CrossRef CAS PubMed.
- D. Moreau, C. Lefort, R. Burke, P. Leveque and R. P. O'Connor, Biomed. Opt. Express, 2015, 6, 4105–4117 CrossRef CAS PubMed.
- R. Fu, B. Xu and D. Li, Int. J. Therm. Sci., 2006, 45, 841–847 CrossRef.
- R. Samy, T. Glawdel and C. L. Ren, Anal. Chem., 2007, 80, 369–375 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ay01758e |
| ‡ The term Spiegelmer is a trademark of NOXXON Pharma AG, Max-Dohrn-Str. 8-10, 10589 Berlin, Germany. |
| This journal is © The Royal Society of Chemistry 2019 |