
A graphene oxide coated gold nanostar based sensing platform for ultrasensitive electrochemical detection of circulating tumor DNA†
Mahbubur
Rahman
 ab,
Daxiang
Cui
*a,
Shukui
Zhou
c,
Amin
Zhang
a and
Di
Chen
a
ab,
Daxiang
Cui
*a,
Shukui
Zhou
c,
Amin
Zhang
a and
Di
Chen
a
aInstitute of Nano Biomedicine and Engineering, Shanghai Engineering Research Center for Intelligent Instrument for Diagnosis and Therapy, Department of Instrument Science & Engineering, School of Electronic Information and Electrical Engineering, Shanghai Jiao Tong University, Shanghai 200240, PR China. E-mail: dxcui@sjtu.edu.cn
bDepartment of General Educational Development (GED), Faculty of Science and Information Technology, Daffodil International University, 102 & 102/1, Shukrabad, Mirpur Road, Dhanmondi, Dhaka 1207, Bangladesh
cDepartment of Urology, Shanghai Sixth People's Hospital, Shanghai Jiaotong University, Shanghai Oriental Institute for Urologic Reconstruction, Shanghai 200233, PR China
First published on 10th December 2019
Abstract
A high-performance electrochemical sensing platform inspired by a functional ‘green’ electrochemical reduction pathway was developed to identify and detect circulating tumor DNA (ctDNA) of gastric carcinoma in peripheral blood. The assembly of the biosensor, consisting of graphene oxide-wrapped gold nanostars (GO-AuNSs), was fabricated on a glassy carbon electrode using the surface chemistry of ‘layer-by-layer’ modification via an electrochemical route. The electrodeposition of the GO-AuNS nanocomposite on the electrode's surface effectively improved its electrochemical response and enhanced its immobilization pathway. At the same time, it demonstrated the electrochemical phenomena caused by DNA immobilization and hybridization events sans complex labelling or other indicators. The immobilization of the probe DNA occurred due to the π–π (weak) interactions between the GO-AuNS composite and the DNA bases. The hybridization of the probe DNA with ctDNA resulted in the formation of a helix structure, which precipitated the release of the resulting dsDNA from the electrode's surface. The immobilization and hybridization events caused a ‘signal off’ and ‘signal on’ model by respectively decreasing and increasing the signal current. The prepared composite nanostructure and the subsequently modified electrodes were characterized using Scanning Electron Microscopy (SEM) and Transmission Electron Microscopy (TEM). The electrochemical response of the biosensor was elucidated using Differential Pulse Voltammetry (DPV), Cyclic Voltammetry (CV), and Electrochemical Impedance Spectroscopy (EIS). A linear relationship was established between the oxidation peak currents (DPV) and the different concentrations of ctDNA within the range of 1.0 × 10−20 M to 1.0 × 10−12 M (R2 = 0.998), with a detection limit (LOD) of 1.0 × 10−20 M (S/N = 3). The developed method shows translational prospects in ctDNA detection.
Introduction
The World Health Organization (WHO) reported that cancer represents the second most prevalent cause of death in the world, accounting for 9.6 million deaths in 2018 alone. Another report postulated that nearly one in six people die of cancer globally.1,2 Genetically predisposed cancer is caused by genetic and epigenetic alterations, which change the gene or protein expression or alter the protein content in a cell. The previously mentioned alteration characteristics are detrimental and can turn into tumoral phenotypes.1 Genomic and environmental aspects (encompassing diets and exposure to harmful elements such as UV rays and other carcinogen factors) are especially influential in the subsequent formation and development of tumors.3 Biosensors/biomarkers are designed to detect specific characteristics (in the form of biological molecules) that are prevalent in cancer cells (such as DNA, RNA, proteins, and their modifications) and also tumors.1 Early detection of cancer represents a challenge to biomedical researchers due to the small window of opportunity present in cancer patients to prevent the proliferation of cancerous cells and eliminate tumors at minimal discomfort and cost.Biosensors and biomarkers have shown a remarkable capability for the early detection of cancer. In the context of early detection and diagnosis of cancerous cells, pinpointing circulating tumor DNA (ctDNA) as a potential biomarker is promising and effective vis-à-vis prognosis.4,5 ctDNA is a prevalent cancer-related biomarker that is present in peripheral blood, representing a variable and minuscule amount of the total circulating DNA. The presence of ctDNA in peripheral blood signifies that the tumor is becoming a burden and progressing towards malignancy.6 These double-stranded circulating DNA fragments carry tumor-specific sequence mutations and are present in the cell-free fraction of blood. The sequence also proves the occurrence of (corresponding and specific types of) cancer, which means that detection methods would require only small amounts of blood for a diagnosis as opposed to inconvenient biopsies.7 The detection of ctDNA is complicated due to its short half-life of less than two hours, while that of other biomarkers is several weeks.8 Another challenge associated with detecting ctDNA is its deficient concentrations and variability in peripheral blood. An effective ctDNA detection method can be used to map the evolution of the cancer cells, which would be useful when informing the patients about the current and probable progression of their condition. The unique characteristics of the ctDNA detection technique as a tool for liquid biopsy makes it useful for specific cancer diagnosis. Usually, ctDNA forms ∼1–0.01% of the circulating DNA in peripheral blood. Its concentration increases at advanced stages of tumor development.4,6 The traditional approaches used for detecting ctDNA in the blood are polymerase chain reaction and DNA sequencing techniques. However, both approaches suffer from reduced sensitivities and suboptimal limits of detection (LODs). Despite their prevalence in ctDNA detection, their widespread use is limited due to complicated sample preparation and inherent interference resulting from biological constituents (environment).9 There are also other techniques that can be used to detect biomarkers, such as those based on enzyme-linked immunosorbent assay (ELISA) and high performance liquid chromatography (HPLC), but these techniques suffer from technological limitations, complex instrumentation, extended detection periods, and costly reagents required for a single detection.9
Biosensors are preferred because they seemingly lack any of the complications associated with detecting biomarkers.8 Gene-based biosensors are becoming more prevalent in the fields of tumor detection, forensic investigation, and environmental monitoring.10–14 One of the more popular tools for gene detection is the electrochemical DNA sensor, which detects oligonucleotide sequences. It is especially useful at tracing unique gene sequences, especially at lower physiological levels15 relative to approaches based on opto-chemiluminescence, the ineffectiveness of the latter owing to the high cost and complexity of the associated equipment required.4 The lower cost and quick turnover of label-free and enzyme-free electrochemical biosensors propelled them to the forefront of DNA detection research.14 An electrochemical biosensor was reported by Chu et al. (2016) for ctDNA detection using thin-layer MoS2/graphene composites synthesized by a hydrothermal method.4 They reported a comparatively narrow linear range of detection of 1.0 × 10−16 M to 1.0 × 10−13 M, with an LOD of 1.0 × 10−17 M. An electrochemical detection approach for mutated ctDNA in lung cancer patients utilizes DNA clutch probes (DCPs) and nanostructured microelectrodes equipped with peptide nucleic acid (PNA) probes that search for unique mutant DNA sequences. A collection of PNA clamps were utilized to obtain an increased specificity allowing distinguishing between mutated DNA sequences from wild-type DNAs and other somewhat similar mutant sequences. This complicated collection of PNA clamp-based electrochemical methods is sensitive up to 1 fg μL−1 of the target mutated sequence within 100 pg μL−1 of wild-type DNAs, which corresponds to a detection of mutations at 0.01% relative to the wild type.16 A PNA based nanoplasmonic biosensor based on localized surface plasmon resonance (LSPR) and the coupling plasmon mode of gold nanoparticles (AuNPs) for ctDNA detection was also reported by A. H. Nguyen et al. in 2015.5 Zhang et al. (2018) reported the fabrication of an electrochemical sensor to detect the circulating tumor DNA within peripheral blood samples with a linear response range of 1.0 × 10−16 M to 1.0 × 10−10 M at an LOD of 1.8 × 10−17 M for a poly-xanthurenic acid-functionalized MoS2 nanosheet detector.8
The development of surface modification engineering and its application in electrochemical detection techniques helps improve the LOD and increase detection sensitivity. Many researchers have utilized sandwich-type nano-structures possessing a high surface-to-volume ratio to immobilize (more) capture probes, which serves to enhance the conductivity and sensitivity of the biosensors. Variations of graphene oxide (GO) and polymer composites were utilized by researchers to improve the electrocatalytic performance of modified electrodes.17,18 The composite on the surface of the electrode acts as a stabilizer for the responses of the electrodes while simultaneously enhancing the electron transfer capabilities of the analytes. Chemically modified graphene-based materials, in the context of redox couples, allow for rapid electron transfer rates due to the presence of intrinsic structural defects. Graphene-based DNA biosensors are known to perform analytically well compared to other more common carbon electrodes.19 The prevalence of two-dimensional (2D) honeycomb lattices on GO-based materials allows for tunable oxygen functionalities on their basal plane. This results in an effective sensing platform for experimental testing of selective detection of bio-materials and biomarkers at higher sensitivities and selectivities.20 Anisotropic nano-graphene synergizes with the substrate causing increased electron mobility, electrical conductivity, and active surface area for analyte binding.21 Highly branched AuNPs differ from other anisotropic nanoparticles, allowing tuning their optical properties via geometrical alterations. AuNSs also provide an excellent platform for in vitro and in vivo diagnostics,22,23 stem cell tracking, bioimaging,24,25 photodynamic and photothermal tumor treatments,26 and immunotherapy.27 Composites containing gold nanostars (AuNSs) and graphene oxides (GO) are some of the most analyzed and used materials in electrochemical sensing.20 AuNSs/graphene composites minimize the restacking and agglomeration of graphene sheets during the reduction process while enhancing selectivity and sensitivity.28
This study proposes an ultrasensitive electrochemical DNA biosensor for the detection of the PIK3CA gene in peripheral blood. The PIK3CA gene available in the peripheral blood of patients with gastric carcinoma is a kind of circulating tumor DNA (ctDNA). A gold nanostar-reduced graphene oxide (AuNS-rGO) nanocomposite was utilized to enhance the electron transfer rates and effective electrochemical responses. A nanocomposite film was deposited onto the glassy carbon electrode (GCE/rGO-AuNS), with the probe DNA directly immobilized onto the nanoplatforms via van der Waals interactions between the bases of a single-strand DNA (ssDNA) and the basal plane of the nanoplatforms.29 The DNA probe was immobilized using a quick and simple carbodiimide reaction (EDC/NHS)30 on the rGO-AuNS modified electrode surface. The immobilized ssDNA probe stands covering the surface on the rGO-AuNS nanocomposite platform and blocking the charge transfer between the nanocomposite platform and the solution, which introduces the ‘signal off’ switching model by decreasing the signal current. A ‘signal on’ switching protocol is caused upon hybridization between the probe ssDNA and the target ctDNA. This results in the release of the dsDNA from the electrode surface, thus decreasing the charge transfer resistance, which increases the signal current. We elucidated changes in behavior caused by the immobilization of DNA and hybridization by directly determining the differential pulse voltammetry (DPV), cyclic voltammetry (CV), and electrochemical impedance spectroscopy (EIS) responses of the guanine bases. Relative to other reported methods, the approach proposed in this work is practical at a lower cost due to the absence of fluorophore labeling and the enzyme amplification step. The proposed biosensor is easier to handle and store, as we do not need to care about photobleaching or enzymatic stability. Its sensitivity is also relatively higher, which puts this approach at a distinct advantage over others.31
Experimental
Chemicals and reagents
1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) and N-hydroxysuccinimide (NHS) were obtained from Sigma-Aldrich (Germany). Gold (Ill) chloride (HAuCl4), trisodium citrate and AgNO3 were purchased from Sigma-Aldrich (Japan). GO nanosheets and ascorbic acid were provided at no cost by Shanghai LingFeng Chemical Reagent Co. Ltd. (China). Potassium phosphate buffer, sodium phosphate buffer, H2SO4, ethanol, NaCl, KCl, and filter membranes (0.22 μm nitrocellulose membranes) were obtained from Shanghai Macklin Biochemical Industries Park, Shanghai, China. Deionized water (18.2 MΩ) was used for cleaning and preparation of the solutions used in this work. All of the chemicals used were of analytical grade.The DNA oligonucleotides of the PIK3CA gene related to gastric carcinoma were synthesized by Shanghai Sangon Bioengineering Limited Company (China). Their base sequences are listed below:
Probe DNA (ssDNA): 5′-AGT GAT TTT AGA GAG-3′;
Complementary DNA (cDNA): 5′-CTC TCT AAA ATC ACT-3′;
One-base mismatched DNA: 
Two-base mismatched DNA: 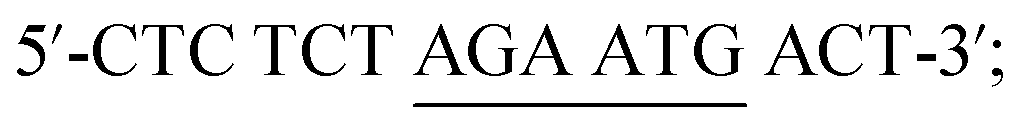
Non-complementary DNA (ncDNA): 5′-TAC TCC GCG CTA ACG-3′.
Synthetic sequence of the E542K oligonucleotide probe of the PIK3CA gene related to gastric carcinoma as the target DNA to investigate the specific complementary ssDNA.
Apparatus
The electrochemical impedance spectroscopy (EIS), differential pulse voltammetry (DPV), and cyclic voltammetry (CV) measurements were performed using a CHI 760E electrochemical workstation (Shanghai CH Instrument Company, China), which includes a glassy carbon electrode (GCE, Ø = 3 mm), an Ag/AgCl electrode, and platinum (Pt) wire as the working, reference, and counter electrodes, respectively. A transmission electron microscope (TEM, Tecnai G2 Spirit Biotwin machine (USA)) and a high-resolution field emission scanning electron microscope (Sirion 200 (FESEM), FEI Company, USA) were used to characterize the samples.Methods
Electrode preparation and electrochemical characterization
The GC electrode's surface (diameter = 3 mm) was sequentially modified using layer-by-layer films (Scheme 1), which were fabricated using cyclic voltammetry (CV). Before the modification, the electrode's surface was cleaned to a mirror finish using aqueous alumina slurries of 0.3 μm and down to 0.05 μm on a polishing pad, after which it was sonicated in deionized (DI) water, ethanol, again in DI water, and finally rinsed with DI water and dried in a N2 atmosphere. | ||
| Scheme 1 Schematic diagram of the fabrication procedure of the electrochemical biosensor for ctDNA detection. | ||
The electrode was electrochemically activated by cycling its potential between −0.2 V and +1.6 V vs. Ag/AgCl at a scan rate of 0.1 V s−1 in 0.5 M H2SO4 until reproducible voltammograms were obtained.32 The roughness factor was calculated from the ratio of the real-to-geometric surface area of the electrode (1.8 ± 0.2) and was kept constant throughout the experiments in this work. The values were determined based on the area of the peak corresponding to the reduction charge associated with the monolayer of chemisorbed oxygen (in the CV obtained in H2SO4). The activated electrode was then characterized by CV (briefly, CV scans were conducted at −0.2 V and +1.6 V vs. Ag/AgCl at a scan rate of 0.1 V s−1 in 0.5 M H2SO4) and EIS in a 2 mM ferri-ferrocyanide solution in 0.1 M KCl. Electrochemical characterization by CV and EIS was also performed after each step of electrode modification.
The rGO-AuNS nanocomposite was electrodeposited onto the GC electrode under deoxygenated conditions (nitrogen bubbling) via a series of 14 cyclic voltammetry cycles between −1.2 and 0.7 V vs. Ag/AgCl at a scan rate of 0.05 V s−1. After the electrochemical reduction of GO and simultaneous electrodeposition of rGO-AuNSs, the modified GCE/rGO-AuNS electrode was rinsed with DI water and then dried in air. The modified electrode was refrigerated at 4 °C for future use. In order to characterize the surface of the electrodes and compare the electrochemical performance, electrodes with and without rGO and AuNSs were prepared. The carboxylic groups were activated by incubating the electrode for 30 min in a mixture of 20 mM EDC and 30 mM NHS. Then, K3[Fe(CN)6] was utilized as the electroactive indicator to monitor the changes occurring in the electrode's surface.
Target ctDNA detection protocol
After incubation in the EDC/NHS coupling reagent, PBS buffer (pH 7.0) containing 10 μL of 1 μM ssDNA capture probe was immediately dropped onto the modified electrode surface (GCE/rGO-AuNS), and then allowed to air dry. The DNA-modified electrode was rinsed thoroughly with DI water to remove the physically adsorbed ssDNA. Then, 10 μL of cDNA (containing PBS, pH 7.0) was pipetted onto the probe-modified GCE/rGO-AuNS/ssDNA electrode and subsequently air-dried. The electrode was kept at a constant voltage of −0.7 V for 300 s to induce its hybridization and release the dsDNA from the electrode's surface. The electrode was afterwards thoroughly rinsed with DI water to remove any unhybridized cDNA from its surface. For the selectivity study, a similar hybridization protocol was utilized to test the (other) probe-modified electrode for hybridization with ncDNA, one-base mismatched DNA, and two-base mismatched DNA.Electrochemical measurements
The EIS measurements were performed in a 2 mM ferri-ferrocyanide solution of 0.1 M KCl with an AC voltage amplitude of 5 mV. The voltage frequencies were within 10 kHz to 0.1 Hz. The CV was measured in a 2 mM ferri-ferrocyanide solution in 0.1 M KCl from −0.6 to +0.2 V at a scan rate of 0.1 V s−1. In order to remove oxygen, a nitrogen gas flow was continuously supplied for the entirety of the experiments. The measurements were repeated three times for each electrode, and the mean values were recorded. All the experiments were conducted at room temperature under atmospheric conditions.Results and discussion
Morphological characterization
The branched gold nanostars (AuNSs) used in this study show a significant optical signal enhancement due to the presence of a highly intense nanoantenna effect from the sharp features of the branches,33–40 which confirms a surfactant-free route for producing biocompatible nanoparticles. The multiple sharp branches (Fig. 1A–C) that protrude from a spherical core are marked as ‘gold nanostars’,37 demonstrating excellent plasmonic properties, two-photon luminescence, and an intense surface-enhanced Raman scattering. It can also be confirmed from Fig. 1A–C that the average size of the AuNSs is ∼80 nm, while the average size of the sharp branches is ∼5–10 nm. | ||
| Fig. 1 Low (A), medium (B) and high resolution (C) TEM images of the AuNSs, and low (D), medium (E) and high (F) resolution TEM images of the rGO-AuNS nanocomposite film. (G) TEM EDS spectrum of the nanocomposite film, and the corresponding elemental mapping data showing the distribution of AuNS nanoparticles on the rGO-AuNS nanocomposite film. (H) XRD spectrum and (I) XPS spectrum of the rGO-AuNS nanocomposite. | ||
Fig. 1D–F show the low, medium and high resolution TEM images of the morphology of the electrochemically deposited ErGO-AuNSs having a smooth surface. The carboxylic group directly links the resulting ErGO-AuNS layers on the GC electrode surface via covalent bonds (C–N). The GO wrapped gold nanostar nanocomposite films overlap and stack on the electrodes' surface.8
Electrode surface modification by electrochemical layer formation
Electrochemical deposition was used to simultaneously reduce the GO and its co-electrodepositant AuNSs to form a GO-AuNS nanocomposite without resorting to reducing reagents on the surface of the electrode. A dispersion was prepared using 0.5 mg mL−1 GO and 2.5 mg mL−1 AuNSs for the electrochemical deposition of the rGO-AuNS nanocomposite under a defined potential range to form an ErGO-AuNS nanocomposite film on the GCE. The formation of the films can be duly confirmed by the presence of a peak of current in the CV cycles (Fig.SI. 3†). The simultaneous reduction of the electrochemically reduced GO (ErGO/rGO)-wrapped AuNS nanohybrid was relatively stable due to its dissolubility in common solvents.20 The AuNSs had an average size of ∼80 nm, and they were homogeneously distributed and wrapped within the rGO film. The characteristics of the composites are not only dictated by the concentration of GO and AuNSs in the solution, but they also rely on the number of deposition CV cycles and scan rates. The increased peak current of the rGO-AuNS deposition remained consistent, which confirms a clear wrapping of AuNSs with the rGO nanosheets.Electrochemical characterization of the GCE/rGO-AuNS nanocomposite electrode
The electrochemical characterization of the bare and modified electrodes was conducted using well-defined CV and EIS techniques in 0.1 M KCl, which included 2.0 mM [Fe(CN)6]3−/4−. A lowest peak current corresponding to [Fe(CN)6]3−/4− was recorded in the case of the bare GC electrode. The peak-to-peak separation (ΔEp) value was determined to be 264 mV, 180 mV, and 86 mV for the bare GCE, GCE/rGO, and GCE/rGO-AuNS electrodes, respectively. After the formation of the rGO-AuNS nanocomposite, a remarkable increase of the redox current and a decreased potential difference became evident. The individual linkages between the AuNSs and the conductive graphene nanosheets resulted in increased rates of electron transfer between the GC electrode surface and its corresponding modifying layers. Furthermore, the fabricated GCE/rGO-AuNS electrode demonstrated excellent electrocatalytic properties.We also determined the electrochemically active surface area (A) for the bare and modified electrodes using the Randles–Sevcik equation.41
| Ip = 2.69 × 105×A × D1/2 × η3/2 × γ1/2C | (1) |
Fig. 2A shows the electrochemical impedance spectrum (EIS) of the differently modified electrodes. The bare GCE has the most massive semicircle with an Rct of 690 Ω (Fig. 2A(a)), which is the lowest transfer rate. The GCE/rGO modified electrode has an Rct value of 385 Ω, as per Fig. 2A(b). The electrode modified by the rGO-AuNP nanocomposite, as seen in Fig. 2A(c), displayed a semicircle diameter that decreased (Rct value of 120 Ω), which could be due to its higher electrical conductivity relative to that of the rGO-only modified electrode, resulting in a higher electron transfer rate in the redox probe.
 | ||
| Fig. 2 (A) Nyquist plots of different electrodes of blank GCE (a), GCE/rGO (b) and GCE/rGO-AuNS composite (c) electrodes, and (B) CVs of the GCE/rGO-AuNS nanocomposite (a), GCE/rGO-AuNS/pDNA (b) and GCE/rGO-AuNS/dsDNA (c) electrodes measured in 0.1 M KCl including 2 mM [Fe(CN)6]3−/4− solution. | ||
The influence of the GO-AuNS nanohybrid on electrochemical detection
The positive electrochemical effectiveness of the GO-AuNSs nanohybrid on the electrochemical detection of ctDNA hybridization can be seen in Fig. 2A. The Nyquist plot of GCE + rGO (Fig. 2A(b)) is smaller than that of the GCE bare electrode seen in Fig. 2A(a), which means that an electroactive layer is present on the surface of the working electrode. The Nyquist plot of the GCE + rGO-AuNS composite seen in Fig. 2A(c) is smaller relative to both the bare and rGO electrodes, which confirms that the rGO and rGO-AuNS nanocomposites positively influenced the electrochemical charge transfer phenomena, as per Fig. 2A.The influence of the GO-AuNS nanocomposite on the electrochemical modification and electrochemical properties of the differently modified electrodes was determined through a comparison of the EIS electrochemical response in 0.1 M KCl including 2 mM [Fe(CN)6]3−/4− solution shown in Fig. 2A. The EIS signal of the GCE/rGO-AuNS composite electrodes was smaller than that of the bare GCE, rGO, and the GO-AuNS nanocomposite, which confirms the positive effect of the electrochemical charge transfer. This could be attributed to the physisorption interactions between the GCE surface and the rich carboxylic group and basal plane of the GO-AuNSs,42 resulting in an increased number of nucleation sites, which ultimately improves the efficiency of the electrodeposition.
The EIS response of the GCE/rGO-AuNS nanocomposite electrode, as per Fig. 2A (curve c), has the smallest Rct value relative to that of the bare GCE and rGO in Fig. 2A (curves a and b), which proves that the prevalence of gold nanostars enhances the electrochemical charge transfer efficiency of the rGO-AuNS nanocomposite.
Optimization of experimental conditions
The final concentration of the GO nanosheets used was 0.5 mg mL−1 and the potential window used was −1.2 to 0.7 V for the electrodeposition of the rGO-AuNS nanocomposite on the GC electrode (Fig.SI. 3†). The optimization of the conditions of the electrochemical CV cycle at −1.2 to 0.7 V is described in Section SI. 4 (Fig. SI. 2†). The loading protocol for the rGO-AuNS nanocomposite was fine-tuned using the EIS responses of the drop-coat and electrodeposition methods; the former resulted in an unstable and poorly conductive nanocomposite while the latter produced a nanocomposite that is the exact opposite. This observation could be attributed to the presence (or lack thereof) of covalent bonds (C–N) in the latter approach (Fig. SI. 1†).Self-signal monitoring of DNA immobilization and hybridization events
The electrochemical performance of the fabricated biosensor was determined using CV in 0.1 M KCl, which includes 2 mM [Fe(CN)6]3−/4− solution post-immobilization and hybridization (Fig. 2B). The biosensor (GCE/rGO-AuNSs) demonstrated excellent electrocatalytic responses within a neutral environment. The probe's DNA was immobilized on the modified surface of the electrode via non-covalent bonding between the rings of the nucleobases and the rGO-AuNS nanocomposite.43 The immobilization resulted in a significant decreased of the self-redox signals of the biosensor (GCE/rGO-AuNSs + pDNA) (Fig. 2B(b)). The immobilized probe DNA acted as a random (and flexible) coil, which could have altered the conformity of the sensor and prevented an effective electron transfer.44The response of the biosensor (GCE/rGO-AuNSs + pDNA) decreased, which is denoted herein as ‘signal-off’. The ssDNA probes were then hybridized with the target ctDNA, which resulted in increased redox signals seen in Fig. 2B(c). Post-hybridization, the resulting dsDNA got released from the surface of the electrode and became associated with the self-signal regeneration of the nanocomposite, in a state regarded as ‘signal-on’ mode.45 Stable hydrogen bonds were formed between the nucleobases and their corresponding shields within the phosphate backbones post-hybridization, resulting in enhanced dsDNA release from the surface of the electrode.46 Overall changes of the redox signal emitted by the rGO-AuNS composite layer confirmed the effectiveness of this biosensor for the direct detection of ctDNA without the need for labels or indicators of hybridization.
Electrochemical detection of complementary target sequences
The electrochemical activity of the modified electrode was analyzed at multiple concentrations of ctDNA using the DPV methods in a full potential window. The GCE/rGO-AuNSs/ssDNA probe was utilized for electrochemical detections of multiple concentrations of complementary sequences. The obtained DPV voltammograms are shown in Fig. 3A. The increasing oxidation currents between pre- and post-hybridization were regarded as the measurement signal. The results confirmed that the oxidation current is linear with the logarithm of the concentration of PIK3CA gene target sequence within 1.0 × 10−20 M to 1.0 × 10−12 M (R2 = 0.998) (Fig. 3B). The detection limit (S/N = 3) was determined to be 1.0 × 10−20 M. | ||
| Fig. 3 DPVs obtained for determination of ctDNA using the GCE/rGO-AuNS electrode in pH 7.4 phosphate buffer at a scan rate of 50 mV s−1 with a wide range of concentrations from 1 × 10−4 μM to 1 × 10−14 μM (A) and the corresponding calibration curves for various concentrations of ctDNA (B). | ||
The broad linear detection range of DNA biosensors can be attributed to the highly monodispersed and spherical rGO-AuNS nanocomposite particles acting as carrier matrices for the purpose of DNA immobilization. The carboxylic acid-rich layer on the rGO-AuNS nanocomposite surface is a suitable binding surface, and its interactions with the DNA capture probes resulted in naturally occurring attachments forming between the constituents. The formation of a nanocomposite using AuNSs with rGO showed a synergistic effect that amplified the analytical signal of the DNA hybridization response, which increased the overall sensitivity of the DNA biosensor.
Reported studies involving modified electrodes vis-à-vis the PIK3CA gene detection displayed detection limits of up to 1.8 × 10−17 M and a detection range of 10−16 to 10−10 M,8 a detection limit of 10−17 M and a detection range of 10−16 to 10−13 M,4 and a detection limit of 50 fM and a detection range of 50–3200 fM.5 These values confirmed the effectiveness of the biosensor for DNA biosensing applications (for a more detailed comparison with other reported studies, see Table 1).
| Methods | Surface modification technique | Target detection | Linear response range | LOD | Ref. |
|---|---|---|---|---|---|
| Electrochemical | rGO/AuNSs nanocomposite on a GCE | ctDNA | 1 × 10−20 M to 1 × 10−12 M | 1 × 10−20 M | This work |
| Electrochemical | Poly-xanthurenic acid/MoS2 composite on a CPE | ctDNA | 1 × 10−16 M to 1 × 10−10 M | 1 × 10−17 M | 8 |
| Electrochemical | Thin layer MoS2/graphene composite on a GCE | ctDNA | 1 × 10−16 M to 1 × 10−13 M | 1 × 10−17 M | 4 |
| Electrochemical | DNA clutch probes (DCPs)/nanostructured microelectrodes equipped with PNA probe clamps | ctDNA | — | 1 fg μL−1 | 16 |
| Electrochemical | AuNP/PNA probes | ctDNA | 50 fM – 3200 fM | 50 fM | 5 |
Selectivity, reproducibility, and stability study of DNA hybridization recognition
The electrochemical impedance spectroscopy (EIS) technique is known to be accurate in its assessment of the interfacial property alteration of the surface of the electrode. Subsequently, the interfacial electron transfer resistance (Ret) can be derived from the diameter of the Nyquist plot.47 The selectivity of the DNA biosensor fabricated in this work was determined using the probe ssDNA to allow it to hybridize with multiple DNA sequences of the PIK3CA gene linked to gastric carcinoma. Changes to the Nyquist plots in [Fe(CN)6]3−/4− post-hybridization of the probe ssDNA are shown in Fig. 4A. The Ret value increased when the probe ssDNA was connected to the GCE/rGO-AuNS nanocomposite, confirming the immobilization of the probe ssDNA. Hybridization with the target ctDNA resulted in a significant decrease in the Ret value. | ||
| Fig. 4 EIS Nyquist plot (A) of the different electrodes measured in 0.1 M KCl including 2 mM [Fe(CN)6]3−/4− solution at the GCE/rGO-AuNSs (a), GCE/rGO-AuNSs/ssDNA (b), GCE/rGO-AuNSs/dsDNA (hybridized with cDNA) (c), hybridized with one-base mismatched DNA (d), hybridized with two-base mismatched DNA (e) and hybridized with ncDNA (f). Oxidation current of DPV measurement obtained from the GCE/ErGO-AuNS electrode measured in 0.1 M KCl including 2 mM [Fe(CN)6]3−/4− solution after storage in a refrigerator at 4 °C (B). | ||
The release of the resulting dsDNA from the nanocomposite surface induced a ‘signal-on’ response on the device. Upon hybridization with the ncDNA, the Ret value (Fig. 4A(f)) showed little change (relative to Fig. 4A(b)), confirming the failure of hybridization. The Ret values are linked to the one-base mismatched DNA (Fig. 4A(d)), while the two-base mismatched DNA (Fig. 4A(e)) exceeded the one in Fig. 4A(c), which means that complete hybridization failed. This failure can be attributed to the previously mentioned base mismatch. The indicator-free DNA biosensor is extremely selective towards the presence (or lack thereof) of the ctDNA hybridization.
The stability of the fabricated DNA biosensor was determined by measuring the current oxidation value of the GCE/rGO-AuNS electrode. The biosensor was refrigerated at a temperature of 4 °C for three weeks. The sensor indicated a ∼3% decrease in its DPV signals after a week, confirming stability within this time period. The DPV signals of the sensors were ∼93% and ∼62% of their respective initial values after 10 and 15 days of storage, respectively, which confirms the stability of the sensors (Fig. 4B).
The reproducibility of the measurements of the fabricated DNA sensor was determined by detecting 1.0 × 10−20 M target ctDNA sequences using five parallel-made probe-modified electrodes. An RSD value of 4.7 ± 0.2% (n = 5) was reported in signal changes, which confirms that the fabricated biosensor is indeed reliable in its measurement capabilities (reproducible).
Conclusions
In summary, a novel electrochemical label-free DNA sensing platform was developed for the detection of ctDNA. The ErGO-AuNS nanocomposite possesses excellent biocompatibility and electrochemical transduction characteristics. Probe immobilization on the DNA was achieved via π–π interactions between the rings of the DNA bases. The resulting conjugated GO-AuNS composite on the surface of the electrode induced a ‘signal-off’ response via charge transfer resistance. Post-hybridization, the resulting dsDNA was released from the GO-AuNS nanocomposite to reduce the charge transfer resistance and cause a ‘signal-on’ response by the biosensor. The fabricated label-free electrochemical DNA biosensor was successfully used to detect the ctDNA of the PIK3CA gene associated with gastric carcinoma. The biosensor demonstrated excellent sensitivity, selectivity, and stability vis-à-vis the target DNA and a wide linear response range of ctDNA concentration of 1.0 × 10−20 M to 1.0 × 10−12 M (R2 = 0.998), with an LOD of 1.0 × 10−20 M. The simple and quick fabrication, label-free detection protocol, low production cost, and detection effectiveness of the GCE/rGO-AuNS nanocomposite make it suitable for use in clinical diagnosis and ctDNA biomedical analysis.Authors' contributions
MR carried out the experiments and drafted the manuscript. DXC supervised the overall study and polished the manuscript. SZ, AZ and DC read and approved the final manuscript.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
This work was supported by the Key Basic Research Program of China (No. 2017YFA0205304), Natural Scientific Foundation of China (No. 81602184 and SKLPBS1827), and Medical Engineering Cross Project of Shanghai Jiao Tong University (YG2016ZD10, ZH2018QNA51, and ZH2018QNA28). This work was also supported by “the Belt and Road” young scientist exchange program of the Science and Technology Commission of Shanghai (Grant No. 18410741600).Notes and references
- A. P. Beltrán and M. García, International Journal of Biosensors & Bioelectronics, 2018, 4, 20–21 Search PubMed.
- J. Ferlay, I. Soerjomataram, R. Dikshit, S. Eser, C. Mathers, M. Rebelo, D. M. Parkin, D. Forman and F. Bray, Int. J. Cancer, 2015, 136, E359–E386 CAS.
- A. Ahluwalia, P. Yan, J. Hurteau, R. Bigsby, S. Jung, T.-M. Huang and K. Nephew, Gynecol. Oncol., 2001, 82, 261–268 CrossRef CAS PubMed.
- Y. Chu, B. Cai, Y. Ma, M. Zhao, Z. Ye and J. Huang, RSC Adv., 2016, 6, 22673–22678 RSC.
- A. H. Nguyen and S. J. Sim, Biosens. Bioelectron., 2015, 67, 443–449 CrossRef CAS PubMed.
- E. Yong, Nature, 2014, 511, 524 CrossRef CAS PubMed.
- H. J. Yoon, T. H. Kim, Z. Zhang, E. Azizi, T. M. Pham, C. Paoletti, J. Lin, N. Ramnath, M. S. Wicha and D. F. Hayes, Nat. Nanotechnol., 2013, 8, 735 CrossRef CAS PubMed.
- W. Zhang, Z. Dai, X. Liu and J. Yang, Biosens. Bioelectron., 2018, 105, 116–120 CrossRef CAS PubMed.
- J. F. Rusling, C. V. Kumar, J. S. Gutkind and V. Patel, Analyst, 2010, 135, 2496–2511 RSC.
- H.-F. Cui, L. Cheng, J. Zhang, R. Liu, C. Zhang and H. Fan, Biosens. Bioelectron., 2014, 56, 124–128 CAS.
- S. Jampasa, W. Wonsawat, N. Rodthongkum, W. Siangproh, P. Yanatatsaneejit, T. Vilaivan and O. Chailapakul, Biosens. Bioelectron., 2014, 54, 428–434 CrossRef CAS PubMed.
- J. Sui, L. Zhang and H. Peng, Eur. Polym. J., 2013, 49, 139–146 CrossRef CAS.
- S. Niu, J. Sun, C. Nan and J. Lin, Sens. Actuators, B, 2013, 176, 58–63 CrossRef CAS.
- M. Rahman, L. Y. Heng, D. Futra and T. L. Ling, Nanoscale Res. Lett., 2017, 12, 474 CrossRef PubMed.
- M. Rahman, L. Y. Heng, D. Futra, C. P. Chiang, Z. A. Rashid and T. L. Ling, Nanoscale Res. Lett., 2017, 12, 484 CrossRef PubMed.
- J. Das, I. Ivanov, E. H. Sargent and S. O. Kelley, J. Am. Chem. Soc., 2016, 138, 11009–11016 CrossRef CAS PubMed.
- G. A. Tığ, J. Electroanal. Chem., 2017, 807, 19–28 CrossRef.
- M. Hasanzadeh, F. Mokhtari, N. Shadjou, A. Eftekhari, A. Mokhtarzadeh, V. Jouyban-Gharamaleki and S. Mahboob, Mater. Sci. Eng., C, 2017, 75, 247–258 CrossRef CAS PubMed.
- A. V. Patil, F. B. Fernandes, P. R. Bueno and J. J. Davis, Bioanalysis, 2015, 7, 725–742 CrossRef CAS PubMed.
- M. Khan, X. Liu, Y. Tang and X. Liu, Biosens. Bioelectron., 2018, 117, 508–514 CrossRef CAS PubMed.
- S. V. Otari, M. Kumar, M. Z. Anwar, N. D. Thorat, S. K. Patel, D. Lee, J. H. Lee, J.-K. Lee, Y. C. Kang and L. Zhang, Sci. Rep., 2017, 7, 10980 CrossRef PubMed.
- H.-N. Wang, B. M. Crawford, A. M. Fales, M. L. Bowie, V. L. Seewaldt and T. Vo-Dinh, J. Phys. Chem. C, 2016, 120, 21047–21055 CrossRef CAS PubMed.
- A. Oseledchyk, C. Andreou, M. A. Wall and M. F. Kircher, ACS Nano, 2017, 11, 1488–1497 CrossRef CAS PubMed.
- S. Harmsen, R. Huang, M. A. Wall, H. Karabeber, J. M. Samii, M. Spaliviero, J. R. White, S. Monette, R. O'connor and K. L. Pitter, Sci. Transl. Med., 2015, 7, 271ra277 Search PubMed.
- C. Andreou, S. A. Kishore and M. F. Kircher, J. Nucl. Med., 2015, 56, 1295 CrossRef CAS PubMed.
- C. G. Khoury and T. Vo-Dinh, J. Phys. Chem. C, 2008, 112, 18849–18859 CrossRef CAS PubMed.
- J. Kneipp, H. Kneipp, A. Rajadurai, R. W. Redmond and K. Kneipp, J. Raman Spectrosc., 2009, 40, 1–5 CrossRef CAS.
- L. Ruiyi, L. Ling, B. Hongxia and L. Zaijun, Biosens. Bioelectron., 2016, 79, 457–466 CrossRef PubMed.
- L. Ruiyi, W. Jiajia, L. Ling and L. Zaijun, Microchim. Acta, 2016, 183, 1641–1649 CrossRef.
- M. Mohiuddin, D. Arbain, A. Shafiqul Islam, M. Rahman, M. Ahmad and M. Ahmad, Curr. Nanosci., 2014, 10, 730–735 CrossRef CAS.
- S. Das, M. Kim, J.-w. Lee and W. Choi, Crit. Rev. Solid State Mater. Sci., 2014, 39, 231–252 CrossRef CAS.
- J. P. Hoare, J. Electrochem. Soc., 1984, 131, 1808–1815 CrossRef CAS.
- A. S. De Silva Indrasekara, S. F. Johnson, R. A. Odion and T. Vo-Dinh, ACS Omega, 2018, 3, 2202–2210 CrossRef CAS PubMed.
- H. de Puig, J. O. Tam, C.-W. Yen, L. Gehrke and K. Hamad-Schifferli, J. Phys. Chem. C, 2015, 119, 17408–17415 CrossRef CAS PubMed.
- F. Hao, C. L. Nehl, J. H. Hafner and P. Nordlander, Nano Lett., 2007, 7, 729–732 CrossRef CAS PubMed.
- C. Hrelescu, T. K. Sau, A. L. Rogach, F. Jäckel and J. Feldmann, Appl. Phys. Lett., 2009, 94, 153113 CrossRef.
- H. Yuan, C. G. Khoury, H. Hwang, C. M. Wilson, G. A. Grant and T. Vo-Dinh, Nanotechnology, 2012, 23, 075102 CrossRef PubMed.
- A. D. S. Indrasekara, S. Meyers, S. Shubeita, L. Feldman, T. Gustafsson and L. Fabris, Nanoscale, 2014, 6, 8891–8899 RSC.
- T. Tsoulos, L. Han, J. Weir, H. Xin and L. Fabris, Nanoscale, 2017, 9, 3766–3773 RSC.
- A. S. D. Indrasekara, R. Thomas and L. Fabris, Phys. Chem. Chem. Phys., 2015, 17, 21133–21142 RSC.
- J. Du, R. Yue, F. Ren, Z. Yao, F. Jiang, P. Yang and Y. Du, Biosens. Bioelectron., 2014, 53, 220–224 CrossRef CAS PubMed.
- X. Wang, W. Ma, T. Ge, T. Yang and K. Jiao, Electrochim. Acta, 2016, 190, 1025–1031 CrossRef CAS.
- S. Liu, L. Wang, Y. Luo, J. Tian, H. Li and X. Sun, Nanoscale, 2011, 3, 967–969 RSC.
- Y. W. Hu, T. Yang, X. X. Wang and K. Jiao, Chem.–Eur. J., 2010, 16, 1992–1999 CrossRef CAS PubMed.
- T. Yang, Q. Guan, X. Guo, L. Meng, M. Du and K. Jiao, Anal. Chem, 2013, 85, 1358–1366 CrossRef CAS PubMed.
- W. Zhang, Z. Dai, X. Liu and J. Yang, Biosens. Bioelectron., 2018, 105, 116–120 CrossRef CAS PubMed.
- A. Bonanni and M. Del Valle, Anal. Chim. Acta, 2010, 678, 7–17 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ay01620a |
| This journal is © The Royal Society of Chemistry 2020 |