
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
1H quantitative NMR and UHPLC-MS analysis of seized MDMA/NPS mixtures and tablets from night-club venues†
Husain A.
Naqi
,
Stephen M.
Husbands
and
Ian S.
Blagbrough
 *
*
Department of Pharmacy and Pharmacology, University of Bath, Bath BA2 7AY, UK. E-mail: prsisb@bath.ac.uk; Tel: +44 (0)1225 386795
First published on 20th August 2019
Abstract
3,4-Methylenedioxymethamphetamine (MDMA) in the UK has increased in purity and the contents sold as MDMA (ecstasy, E) have increased in complexity. Night-club scenes remain overall the biggest venues where such powders or tablets are consumed. HPLC and GC are the gold standard analytical methods in forensic laboratories for the quantification of seized samples of illicit drugs. However, complex mixtures of such samples may be a limiting factor for chromatographic techniques. NMR is used for the structural elucidation of newly isolated natural products or synthesized compounds, but also the inherent ability of 1H NMR to quantify compounds is a powerful tool that is employed for quantification and purity testing in the pharmaceutical industry. In this study, a 1H quantitative NMR (q-NMR) method is developed for the quantification of seized samples from night-clubs. These samples are shown to contain mixtures of MDMA and other NPS, e.g. ethylone, methylone, trifluoromethylpiperazine (TFMPP), N,N-dimethyl-3,4-methylenedioxyamphetamine (MDDMA), 4-bromo-2,5-dimethoxyphenethylamine (2C-B), and 4-iodo-2,5-dimethoxyphenethylamine (2C-I). The method is applied to MDMA tablets seized from similar venues, and compared with UHPLC and UHPLC-MS, resulting in a good agreement across techniques. 1H q-NMR provides a fast (15 min) and robust analytical method for the quantification of complex seized samples without resorting to tedious sample preparation or obtaining reference standards.
Introduction
3,4-Methylenedioxymethamphetamine (MDMA, ecstasy, E) is considered to be by far the most popular of the phenethylamines in Europe, as demonstrated by the amount seized, 5.3 million tablets and 295 kg of powder in 2016.1 This popularity is the result of many factors such as the high purity (higher than seized amphetamine and methamphetamine) and the low price of only 6–11 euros/tablet.1 The health risks of high-potency products and the continued emergence of new substances, together with the changing patterns of drug use are among the issues highlighted in the European Drug Reports of 2016![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 2 and 2018,1 and recently further reinforced in the June 2019 Report.3
2 and 2018,1 and recently further reinforced in the June 2019 Report.3
Cathinones are the second most popular class by seizures among novel psychoactive substances (NPS) (after synthetic cannabinoid receptor agonists, SCRAs) and the leading NPS available in the powdered form.1 They are based on the natural product S-cathinone, with many opportunities in the chemical structure for modifications with e.g. alkyl substituents resulting in a large number of derivatives, e.g. methylenedioxy substituents giving rise to analogues such as butylone, ethylone and methylone.4,5 The pharmacology and dose of cathinones and MDMA are of importance, especially in the context of analytical and forensic toxicology. MDMA works primarily on serotonin receptors (5-HT-R), but also possesses dopamine release action.4,5 Mephedrone is a more potent inducer of locomotor activity in rodents than MDMA.4 Therefore, taking both types of drug can lead to serious consequences such as tachycardia, hypertension, hyperthermia and dehydration.6,7 The association of cathinones with each other and with other NPS has been described by Zuba and Byrska8 Using LC, GC-MS and NMR, they showed that the most common cathinones detected were 3,4-methylenedioxypyrovalerone (MDPV) and butylone, found with piperazines. Toxicological identification of post-mortem cases showed MDMA with methylone, and MDMA with ethylone.9,10
NPS continue to increase in number, e.g., 101 NPS were discovered for the first time in 2014. They can be toxic, as the taken dose is not known, and occasionally they have fatal consequences, often extensively reported in the media. MDMA trafficking continues to grow in complexity and tablets are commonly sold containing different doses of MDMA, often mixed (cut) with other NPS. Therefore, identification and quantification of drugs of abuse is still a challenge. New detection strategies and reports towards providing point-of-care/in-the-field NPS sensors have been reviewed.11
The simultaneous detection of substances present in drugs of abuse is increasingly important as some materials are known for their high mortality rate. One drug that has received considerable attention is p-methoxyamphetamine (PMA), commonly known as “Dr Death”. This amphetamine is linked with several deaths internationally and is often mixed together with MDMA, but still sold as “ecstasy”. Banks, Sutcliffe and co-workers at Manchester Metropolitan University have reported the simultaneous detection and quantification of MDMA and PMA through an electrochemical technique using screen-printed graphite electrodes (SPEs). This electrochemical analysis was shown to be an improvement over presumptive colour tests which were found not to be able to discriminate when MDMA and PMA were both present in the sample. Their novel electrochemical protocol was independently validated in a synthetic MDMA/PMA sample with HPLC.12 The continuous and progressive increase in the adulteration of common illicit street drugs causes overdoses, sometimes with fatal consequences. The need for the development of sensitive, selective and reliable analytical protocols for their separation and quantification is being addressed by the simultaneous electrochemical (amperometric) detection using a commercially available impinging jet flow-cell that incorporates in-house SPEs demonstrating high sensitivity and reproducibility for the analysis of illicit drugs of abuse in the presence of common adulterants (e.g. caffeine, paracetamol and benzocaine) and co-formulated excipients (starch, lactose, aerosil 200, etc.) simultaneously electroanalytically sensed within seized street samples,13 and also capable of detecting drug, e.g. mephedrone, metabolites.14
There are other analytical approaches, but each has their limitations of course. Quantification of the contents in commonly ab/used MDMA tablets has been developed using near infrared spectroscopy (NIR) in transmission mode and shown to be more suitable than in diffuse reflectance. The seized MDMA samples were shown also to contain amphetamine and N-ethyl-3,4-methylenedioxyamphetamine (MDE) in different concentrations. This NIR analytical method was referenced to HPLC with diode array detection (DAD).15
Most of the analytical methods used for the quantification of MDMA tablets and cathinones are chromatographic techniques that may be coupled to MS.11 Even though chromatography is the gold standard in industrial and forensic laboratories for quantitative analysis, it suffers from disadvantages.16 Lengthy method development and preparation of mobile phases are time consuming. Furthermore, UHPLC is unable to detect impurities that have no chromophore or are adulterated with high polarity excipients such as glycerol and sugars. Another disadvantage is the poor ionization of such excipients in MS analysis. GC-MS analysis suffers from the absence of or only a weak mass ion, and requires derivatization for amphetamine-type stimulants (ATS) in order to achieve accurate quantitative results.17
On the other hand, NMR requires no solvent preparation, no method development and no stationary phase (column) with which the compounds will interact.16,18 NMR allows a simultaneous structure elucidation and quantification of the targeted analyte in a relatively fast time compared to various different chromatographic techniques and NMR will detect organic impurities, even ones not possessing a chromophore. NMR, despite being a powerful analytical technique, has been underutilized for the detection and quantification of illicit drugs in mixtures. NMR can simultaneously perform the identification and quantification of other substances also present in MDMA tablets, adulterants, that remain a challenge in forensic drug analysis. NMR spectroscopic analysis therefore provides a great opportunity for the characterization and quantification of complex samples containing multiple components. With the development of high-field NMR instruments, sensitivity is improving and that is reflected in quantitative NMR experiments with lower amounts of analyte. 1H NMR is inherently quantitative, as the intensity of the signal being integrated is directly proportional to the number of protons represented by the signal, with the exception of some exchangeable protons (OH, NH, SH). However, this is only true when certain parameters are met such as the relaxation delay (T1) of the signal which can be measured using an inversion recovery pulse sequence of the signals of the analyte. Leaving 5 × T1 to recover the magnetization to 99.3% of its size allows accurate integration for quantitative results, given an appropriate number of scans to provide an acceptable signal-to-noise (S/N) ratio.18,19
Hays has reported a rapid, sensitive, accurate, precise, reproducible, and versatile method for determining the purity of illicit drugs and adulterants using 1H NMR spectroscopy against a high purity internal standard (IS).16 The NMR experiment employs only 8 scans using a 45 s delay and 90° pulse. For quantitation, the chosen NMR signals must be baseline resolved. The relative standard deviation (RSD) of these signals is usually <1% for pure standards, and the results agree well with other purity determining methods. Typically the analyte is dissolved in D2O with maleic acid (MA) as the IS (5 mg) for a range of concentrations from 0.033 to 69.18 mg mL−1 with a resulting correlation coefficient of >0.9999.16
Similarly, in independent studies, Maldaner, de Oliveira and co-workers quantified MDMA by GC-FID and 1H q-NMR (at 600 MHz) also using MA as the IS.20 NMR was shown to be more efficient and versatile than GC in accomplishing the identification and quantification of target analytes in a single analysis with excellent results of accuracy (relative error < 5%) and precision (relative standard deviation, RSD < 2%). Another strength of the NMR method is that it does not require a specific reference material for analysis. In their research, 38 different seized MDMA tablet batches were analysed by GC and q-NMR. Seized tablets weighed between 158–430 mg and five different excipients were identified by FTIR analysis: cellulose – found in 60% of the batches analysed, sucrose, starch, talc, and esters of long chain fatty acids. As well as MDMA, at least one adulterant from: aminopyrine, caffeine, procaine or amphetamine was identified in 6/38 of the analysed samples. The MDMA·HCl purity ranged from 10 to 77%, a mass of 39–152 mg per tablet.20
Recent research has led to the rapid identification of NPS, including MDMA, using a low-field (LF) (60 MHz) benchtop 1H NMR spectrometer.21 Indeed, the screening, detection and quantification of illicit drug “spice” samples using LF NMR spectroscopy has been explored by Gilard and co-workers showing that the recent introduction of benchtop cryogen-free LF NMR spectrometers (60 MHz) can provide useful insights, in the form of diagnostic signals, to chemical structure.22 However, the greater spectral band overlap compared to data acquired from a superconducting magnet NMR spectrometer did present a data analysis challenge.23,24
In this study, the quantitative analysis of multicomponent MDMA with other NPS, such as cathinones, phenethylamines, and piperazines, will illustrate the complexity of the samples from UK night-club venues. Additionally, a cross-method confirmation of q-NMR using an IS method is used to assay different MDMA tablets seized from night-club venues in Bristol. Validation is by UHPLC and UHPLC-MS using MDMA-d5 as an IS for the latter.
Experimental section
Chemicals and sample preparation
NMR grade solvents D2O 99.9% and TMSP-2,2,3,3-d4 98.0% were purchased from Cambridge Isotope Laboratories (Goss Scientific, UK). Methanol and water for UHPLC and UHPLC-MS analysis were Lichrosolve Honeywell LC-MS grade. ±−MDMA 1.0 mg mL−1 in methanol solution reference standard, ±−MDMA-d5 1.0 mg mL−1 in methanol solution reference standard, maleic acid (MA) 99.94% and dimethyl sulphone (DMS) 99.96% are TraceCERT certified for quantitative NMR analysis, and acetanilide (99%) were purchased from Sigma-Aldrich (UK).Drug samples were provided by the Drug Expert Action Team (DEAT), Avon and Somerset Constabulary, in sealed evidence bags containing numerous drug samples in different forms (capsules, crystals, powders, tablets and plant materials) in different packaging (small packs, magazine twists). Samples in small packs in the form of crystals and powders weighing between 50–250 mg were from different night-club venues in Bristol, while MDMA tablet weights ranged between 188–645 mg. For quantitative analysis, crystals and powders were weighed using a Sartorius analytical balance MC 5 followed by extraction with D2O containing IS maleic acid (2.0 mg mL−1) and 0.5% of TMSP-2,2,3,3-d4 (1.0 mL) for NMR spectroscopic analysis.
For MDMA quantitative analysis, tablets of different sizes and shapes were photographed for documentation, followed by manual pulverization using a mortar and pestle. 10.0 mg of powder was weighed using a Sartorius analytical balance MC 5, transferring into a 7.0 mL glass screw neck specimen vial (Fisher Scientific), then extraction with D2O (2.0 mL) containing maleic acid (1.0 mg mL−1) as an NMR quantitative IS with sonication for 30 minutes, filtration through a Sartorius Minisart® 0.2 μm filter followed by taking a final volume of 1.0 mL for NMR spectroscopic analysis. For UHPLC analysis, the sample was diluted 100-fold into a MS vial. Samples were quantified using a 6-point calibration curve 1.5, 3.1, 6.3, 12.5, 25.0, and 50.0 μg mL−1 prepared in UHPLC solvent. For UHPLC-ESI MS quantitative tablet analysis, the sample was extracted with LC-MS grade water and diluted 10000-fold into a MS vial and spiked with MDMA-d5 (500 ng mL−1) as an IS. Samples were quantified using an 8-point calibration curve from 32.25, 62.50, 125.0, 250.0, 500.0, 1000, 2000, and 4000 ng mL−1. Each concentration was spiked with IS MDMA-d5 (10 μL of 25 μg mL−1) to achieve a final IS concentration of 500 ng mL−1. The response was calculated as the ratio of the area under the curve of the target compound to that of the IS.
Instrumentation
Results and discussion
In this study the association (formulation) of MDMA with other NPS is presented. Quantification using a fast, simple and accurate 1H q-NMR of each of the components without resorting to tedious preparation and method development assays will be applied to complex samples. Additionally, seized MDMA tablets were quantified using 1H q-NMR and cross-method confirmed using UHPLC equipped with a variable wavelength detector (VWD) and UHPLC-MS using MDMA-d5 as an internal standard (IS). Eqn (1) was used for 1H q-NMR quantitation: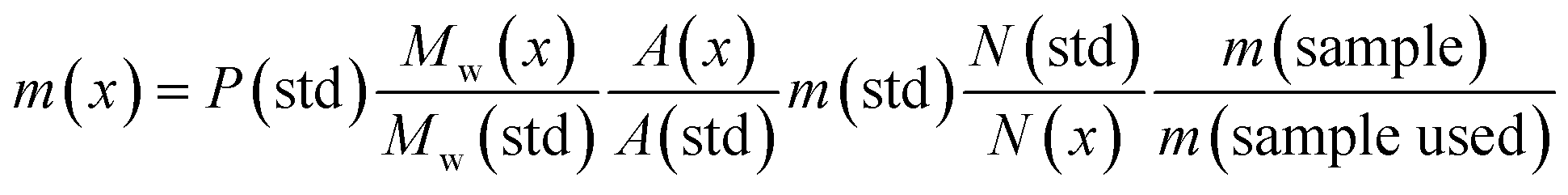 | (1) |
1H q-NMR of MDMA/methylone/trifluoromethylpiperazine (TFMPP) and MDMA/ethylone mixtures using maleic acid (MA) as an IS resonating at δ = 6.38 ppm was performed (Table 1). MA is an ideal IS due to its simple resonance signal (singlet), non-overlapping peak, high purity (99.94%) and high solubility in D2O. An inversion recovery NMR experiment of MDMA, methylone and TFMPP in the mixture was performed to establish the relaxation time (T1) of the signals selected for quantification, to satisfy the parameter of adequate relaxation delay. This was found by experiment to be between 1.5–4.4 s, adopting at least 5 × T1 to ensure complete relaxation of the signals between pulses.16,18,19 Additionally, no more than 10.0 mg of sample was used for quantitative NMR analysis in D2O as the high salt concentration affected the broadness of the peaks by affecting the shimming.
| Sample name | Components identified | mg/10 mg of sample ± SD | RSD% |
|---|---|---|---|
| HN26 n = 6 | MDMA | 1.99 ± 0.2 | 6.4 |
| Ethylone | 6.54 ± 0.1 | 0.9 | |
| HN31 n = 6 | MDMA | 5.01 ± 0.1 | 0.3 |
| Ethylone | 3.40 ± 0.2 | 5.5 | |
| HN48 n = 3 | MDMA | 5.85 ± 0.1 | 1.2 |
| Ethylone | 2.94 ± 0.3 | 11.0 | |
| HN153T n = 6 | MDMA | 3.22 ± 0.2 | 5.6 |
| Methylone | 2.31 ± 0.2 | 7.2 | |
| TFMPP | 2.50 ± 0.1 | 3.8 | |
| HN154 n = 6 | MDMA | 7.42 ± 0.3 | 4.2 |
| Ethylone | 0.71 ± 0.1 | 5.1 | |
| HN157 n = 6 | MDMA | 7.08 ± 0.2 | 2.4 |
| Methylone | 0.53 ± 0.1 | 8.2 | |
| TFMPP | 0.18 ± 0.1 | 7.5 |
In this study, quantitative analysis of different selected samples of MDMA/NPS powder and crystal mixtures was achieved, with simple extraction using D2O containing MA (2.0 mg mL−1) as an IS (Table 1).16,20 Initially the samples were identified and characterized using 1D/2D NMR and LC-ESI/MS. Structural elucidation allowed the selection of signals for quantification of each component with high confidence. The amounts of MA and TMSP-d4 were fixed throughout the analyses. The MA olefinic signal (at δ = 6.38 ppm) was used for quantification. It is well separated from the rest of the components in the mixture, even the methylenedioxy peaks of MDMA, ethylone and methylone, and was normalized to the value of 1.00. TMSP-d4 not only allowed the referencing of the NMR spectra to 0.00 ppm, but also was used to monitor the ratio of MA/TMSP in case of any impurity appearing beneath the MA peak, especially any cutting agents in the maleate form.
In the MDMA/methylone/TFMPP mixtures, the same signal overlapped with the 1′ and 2′ of TFMPP piperazine ring (Fig. 1), whose signals were also excluded from the integration/quantitation analyses. In all the mixtures, all the signals of MDMA were available for integration with the exception of the chiral centre signal (2) at 3.39 ppm, where in MDMA/ethylone mixture an unknown impurity resulted in a significantly higher integration value compared to the rest of the signals (Fig. 2). Ethylone signals for integration depended on the signal to noise (S/N) ratio. In samples HN26, 31 and 48, all the signals were of an appropriate S/N, while in sample HN154, low S/N allowed only the aromatics and methylenedioxy signal (7′) to be integrated. The rest of the signals were too close in proximity to MDMA signals. In methylone spectra, each signal was used for quantitation except the 1′′ methyl-amine signal at 2.69 ppm due to its close proximity with the MDMA 13C satellite signals of 1′′ and 1.
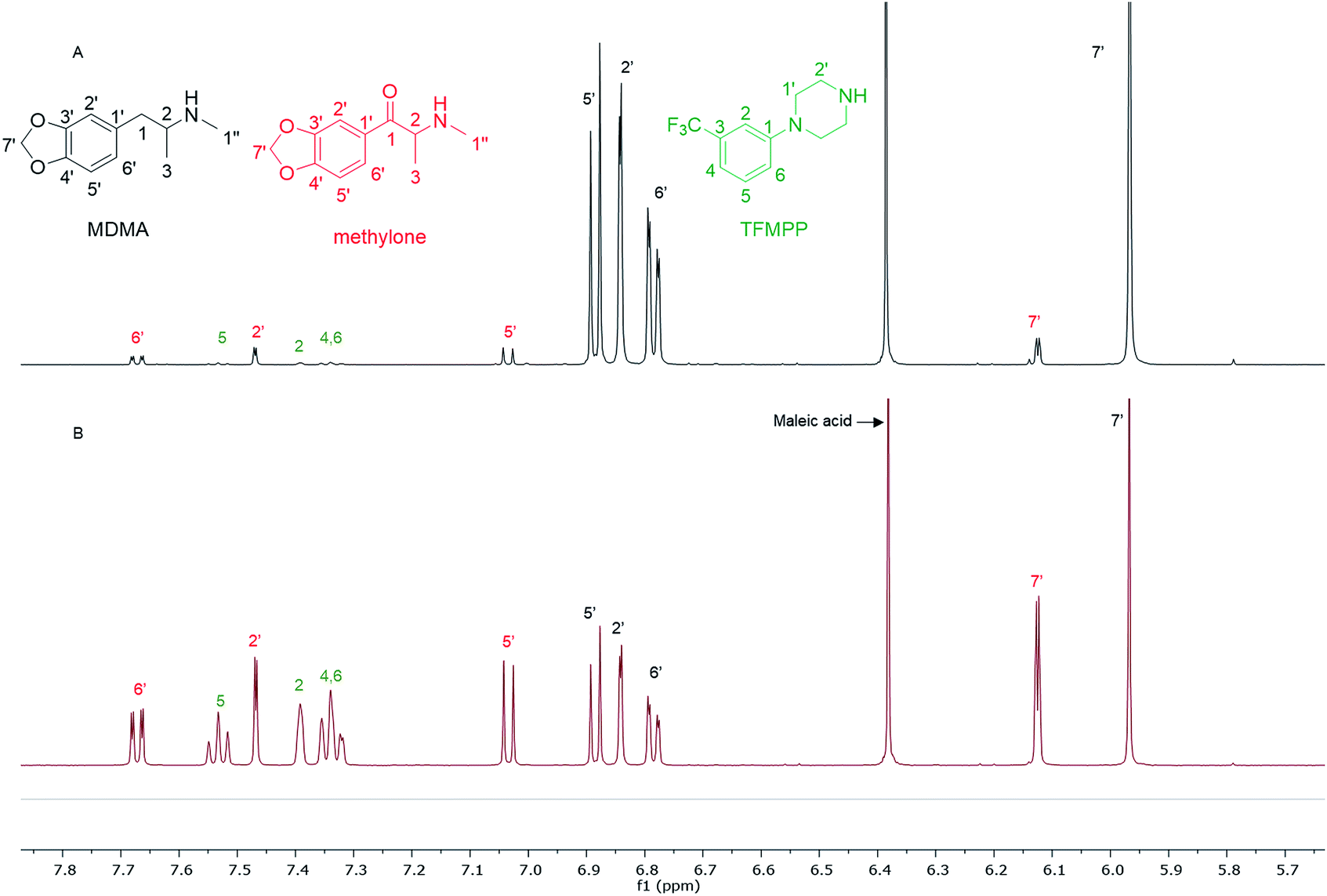 | ||
| Fig. 1 Spectra of 1H NMR (in D2O) between 5.70–7.80 ppm of (A) sample HN157 and (B) sample HN153T, revealing significant differences in the ratio of each component. | ||
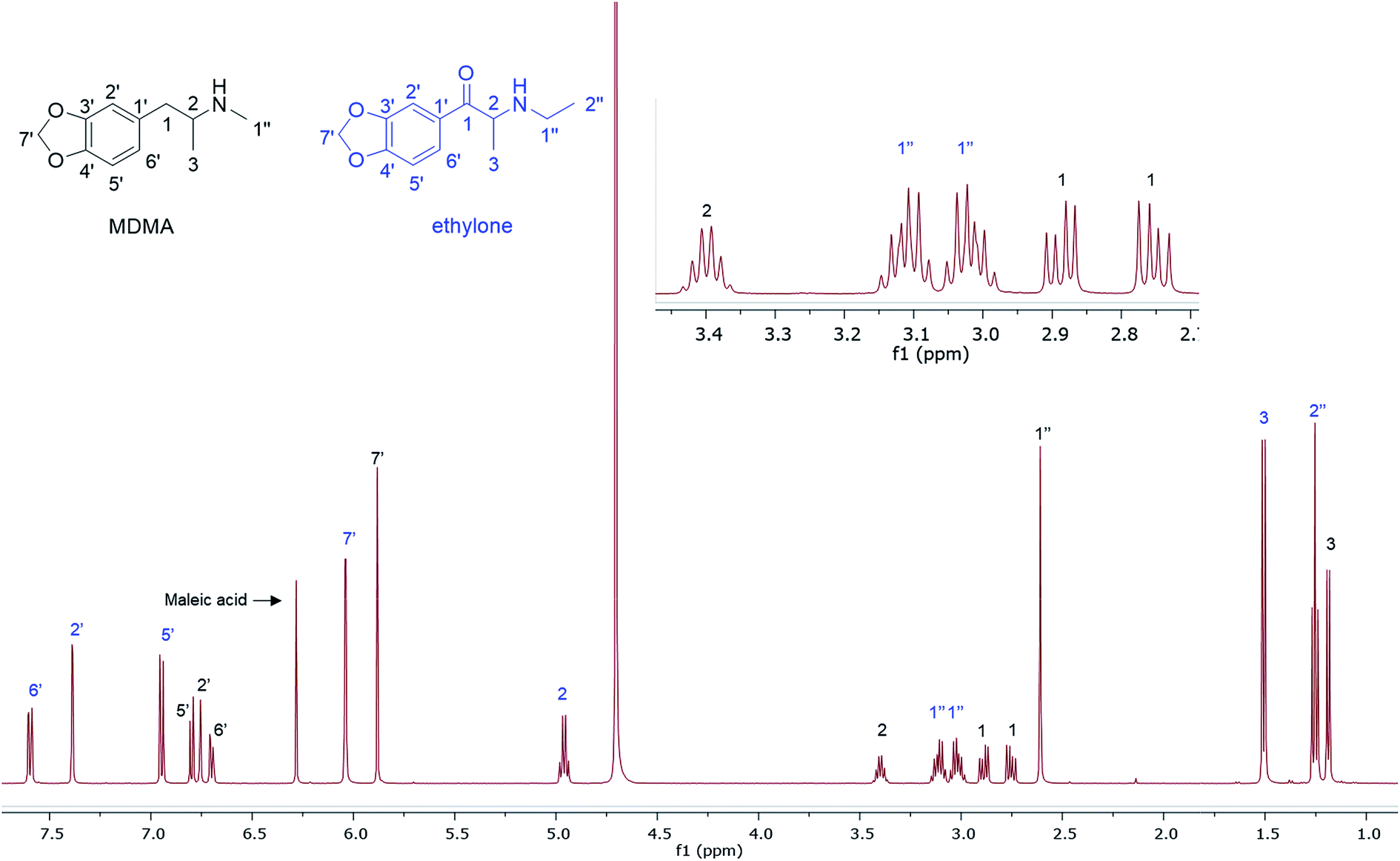 | ||
| Fig. 2 1H NMR (500 MHz in D2O) of MDMA/ethylone mixture with MA as a quantitative IS. | ||
The RSD% is affected by the non-uniformity of the original sample, and even though homogenization was carried out on the powder material some components comprising less than 30% of the sample, and especially less than 10% of the sample, resulted in more than 5% RSD.25 An example of this is the minor components of sample HN157, TFMPP and methylone compared to the major component MDMA (Table 1 and Fig. 1). Furthermore, the total composition of the samples is between 77.9%–87.9%. This is possibly due to the presence of insoluble materials, moisture in the sample, and even an excess of the salt to drug ratio in illicit drug samples as explained by Hays, in which salt and water content have influenced the purity of the samples under investigation by 1H NMR.16 Two important parameters were taken into consideration: the S/N ratio was more than 150, and the integration. The latter is believed to be the most significant parameter for error introduction because of it being operator dependent. Therefore, consistency in integration across all the samples is crucial for obtaining accurate quantitative results.26,27
A fast (15 min), accurate and reproducible 1H q-NMR analysis was performed on many samples (n = 33) containing complex mixtures of MDMA and NPS. The extraction step with D2O was compatible with all the components present in their salt forms due to their high water solubility. NMR allowed the separation of the majority of peaks, the few overlapping signals were excluded from the analysis. The RSD% affected the minor components (<10%) in the samples possibly due to the non-uniformity of the samples. This q-NMR analysis is suitable for an NPS where there is no reference standard available for chromatographic analysis. The analysis results revealed a mixing-trend rather than cutting MDMA with NPS in different ratios. Mixing is perhaps experimentation to increase the pharmacological effect. Cutting is dilution to increase profit. It is outside the scope of this analytical research to determine how such samples are marketed when 2 (or more) illicit compounds are mixed.
26 tablets belonging to 11 different brands of MDMA seized from different dance venues in the Southwest of England were quantified by 1H q-NMR and confirmed by UHPLC using a Variable Wavelength Detector (VWD) and UHPLC-MS (Table 2). For UHPLC analysis, the non-specific (in terms of analyte) UV wavelength of 210 nm was selected to detect impurities present in tablets.
| No | MDMA tablet | Sample | Weight (mg) | UHPLC | NMR | ||
|---|---|---|---|---|---|---|---|
| MDMA dose (mg) ± SD | RSD% | MDMA dose (mg) ± SD | RSD% | ||||
| 1 |

|
a | 468.77 | 82.80 ± 3.0 | 3.6 | 89.08 ± 0.5 | 0.6 |
| b | 494.71 | 108.65 ± 0.9 | 0.8 | 105.26 ± 0.9 | 0.8 | ||
| c | 486.26 | 93.99 ± 1.7 | 1.8 | 97.16 ± 0.8 | 0.8 | ||
| 2 |

|
a | 449.57 | 69.76 ± 1.0 | 1.4 | 76.24 ± 0.3 | 0.4 |
| b | 449.11 | 75.64 ± 1.1 | 1.5 | 81.54 ± 0.3 | 0.4 | ||
| c | 443.17 | 72.94 ± 2.2 | 3.1 | 75.00 ± 1.1 | 1.5 | ||
| 3 |

|
a | 520.37 | 121.13 ± 0.6 | 0.5 | 121.24 ± 0.7 | 0.6 |
| b | 477.08 | 113.26 ± 0.4 | 0.3 | 112.66 ± 0.5 | 0.4 | ||
| c | 501.74 | 94.38 ± 0.6 | 0.6 | 92.58 ± 0.8 | 0.9 | ||
| 4 |

|
a | 512.73 | 160.55 ± 0.8 | 0.5 | 164.21 ± 0.4 | 0.3 |
| b | 481.95 | 150.27 ± 2.4 | 1.6 | 151.92 ± 2.1 | 1.4 | ||
| c | 496.73 | 160.01 ± 0.5 | 0.3 | 165.13 ± 0.5 | 0.3 | ||
| 5 |

|
a | 513.82 | 162.88 ± 3.0 | 1.8 | 163.45 ± 0.3 | 0.2 |
| b | 520.66 | 160.95 ± 3.6 | 2.2 | 165.10 ± 0.9 | 0.5 | ||
| c | 514.93 | 166.54 ± 1.5 | 0.9 | 171.55 ± 1.4 | 0.8 | ||
| 6 |
|
a | 325.00 | 89.45 ± 0.2 | 0.2 | 89.08 ± 1.2 | 1.4 |
| 7 |
|
a | 233.1 | 87.79 ± 0.6 | 0.6 | 90.98 ± 0.2 | 0.2 |
| 8 |

|
a | 211.12 | 85.63 ± 0.1 | 0.1 | 88.36 ± 0.7 | 0.8 |
| b | 209.21 | 83.31 ± 0.3 | 0.4 | 84.18 ± 0.3 | 0.4 | ||
| c | 187.27 | 78.38 ± 0.8 | 1.0 | 81.55 ± 0.5 | 0.6 | ||
| 9 |

|
a | 503.50 | 147.70 ± 2.0 | 1.3 | 151.77 ± 3.2 | 2.1 |
| b | 468.77 | 139.74 ± 1.7 | 1.2 | 144.66 ± 1.4 | 1.0 | ||
| c | 476.81 | 155.02 ± 3.2 | 2.1 | 153.75 ± 1.2 | 0.8 | ||
| 10 |

|
a | 644.70 | 87.13 ± 0.9 | 1.0 | 95.08 ± 0.4 | 0.5 |
| 11 |
|
a | 273.54 | 85.34 ± 1.8 | 2.2 | 87.39 ± 0.7 | 0.8 |
| b | 288.10 | 91.57 ± 0.7 | 0.8 | 88.59 ± 1.5 | 1.7 | ||
Both UHPLC using UV and UHPLC-MS quantification were determined first by determining the % of MDMA, followed by calculating the MDMA content using eqn (2) and (3):28
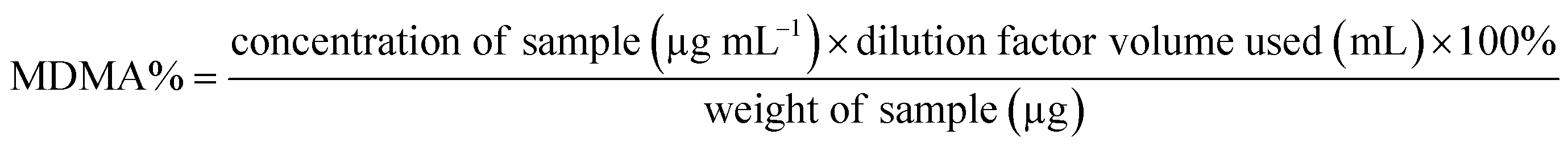 | (2) |
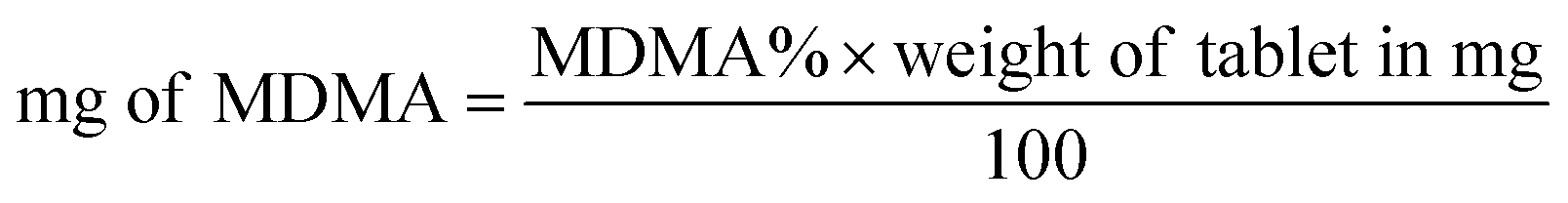 | (3) |
A single extraction, using D2O for NMR analysis and H2O for UHPLC and UHPLC-MS analysis, was employed. This has been deemed sufficient with 100% recovery according to the literature.20,28 Additionally, numerous tablets were qualitatively analysed for cutting agents and types of excipients using NMR. The UHPLC method gave a good linearity on plotting the concentration (μg mL−1) against UV response (Fig. 3). Using TFA provided a better buffering capacity for MDMA, with good peak resolution and no peak tailing compared to using FA with UV detection.
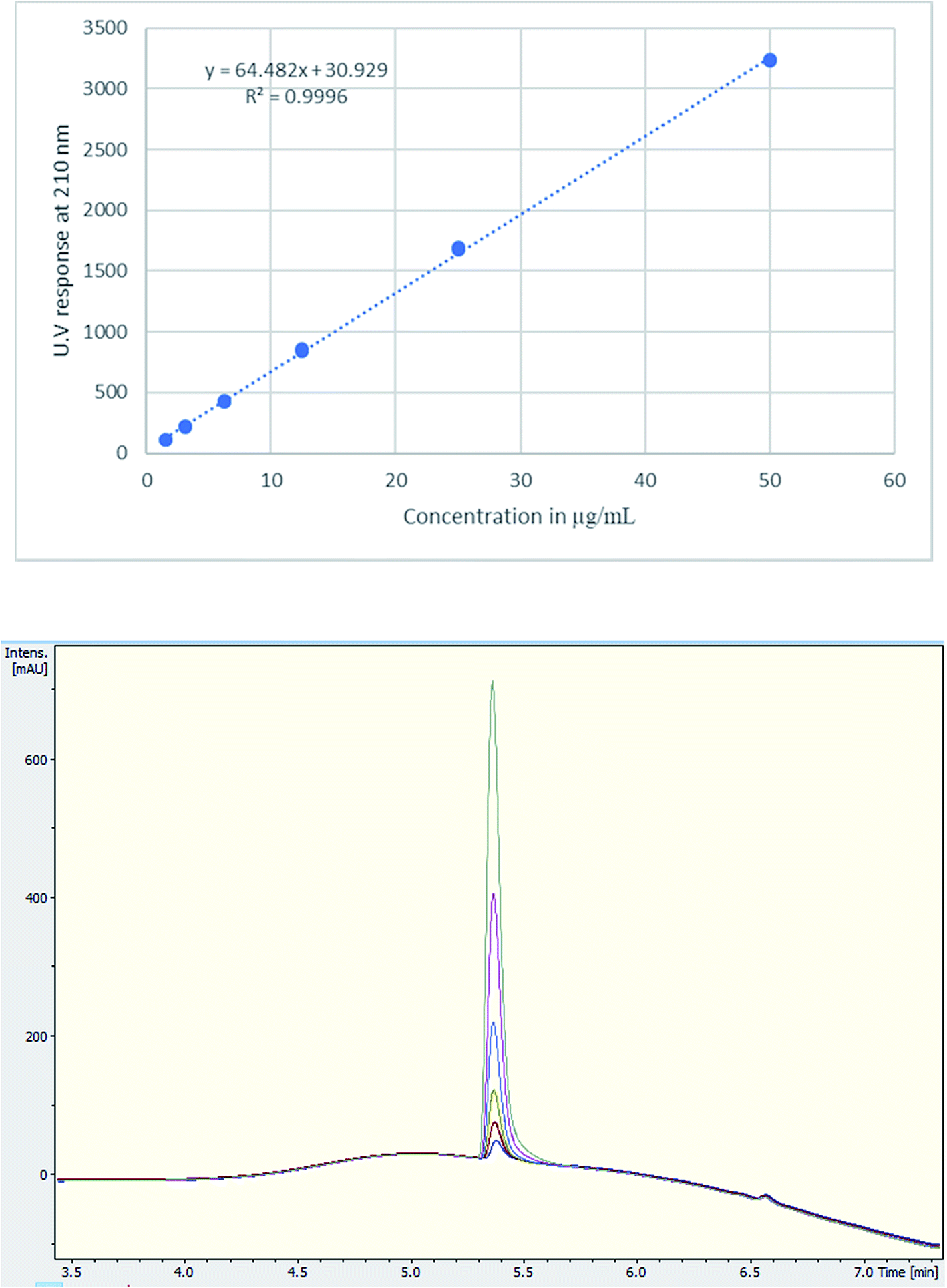 | ||
| Fig. 3 (Upper) Calibration curve of MDMA between 1.5–50 μg mL−1, R2 = 0.9996, (lower) UV chromatogram MDMA RT = 5.4 min of the calibration curve concentrations. | ||
For UHPLC-MS, two tablets from the red UPS brand were selected for comparative analysis (Table 3). The calibration curve was run in triplicate, and gave a good linearity by plotting the concentration (ng mL−1) against response ratio of MDMA to MDMA-d5. The Extracted Ion Chromatograms (EIC) (Fig. S1†) and spectra (Fig. S2†) of the molecular ions of both MDMA and MDMA-d5 were used for quantification. Using a deuterated analogue as an IS is optimal, due to it satisfying the IS criteria by having the same chromatography yet with a different mass ion, no possibility of it existing in the targeted analyte, and possessing similar ionization in ESI MS.29,30 Furthermore, the RSD% across all methods gave a good RSD% of an acceptable value lower than 5% for powdered samples as set out by the European Network of Forensic Science Institute,25 with the exception of one sample 4b using UHPLC-MS analysis. The results of the UHPLC and NMR analyses were comparable. Additionally, using ANOVA single factor analysis of the three methods (NMR, UHPLC, and UHPLC-MS), there is no significant statistical difference (p > 0.05) (Table 3).
| No | UHPLC | NMR | UHPLC-MS | |||||
|---|---|---|---|---|---|---|---|---|
| 4 | Entry | Weight (mg) | MDMA dose (mg) ± SD | RSD% | MDMA dose (mg) ± SD | RSD% | MDMA dose (mg) ± SD | RSD% |

|
a | 512.73 | 160.55 ± 0.8 | 0.5 | 164.21 ± 0.4 | 0.3 | 161.07 ± 4.7 | 2.9 |
| b | 481.95 | 150.27 ± 2.4 | 1.6 | 151.92 ± 2.1 | 1.4 | 153.80 ± 8.6 | 5.6 | |
MA was selected as a quantitative IS due to its high solubility in D2O, simplicity of the olefinic signal (singlet), non-overlapping signal at δ = 6.38 ppm. Due to the hygroscopic nature of MA, the purity was checked against high purity DMS in D2O and acetanilide in DMSO-d6. TMSP-d4 was used for referencing at 0.00 ppm using a fixed concentration of 0.5%. This allowed the use of the ratio of MA/TMSP-d4 to check for any impurities that might be under the MA peak, such as drugs in the maleate salt form. Prior to commencing the analysis, an inversion-recovery experiment was carried out to establish the time it takes the signals of MDMA to relax which ranged between 2–4 s. For MA and DMS, the relaxation times were 6.1 s and 2.9 s respectively.31 Based on these T1 values, the relaxation delay of the pulse sequence was set to achieve at least 5 × T1 for almost complete relaxation.18,19
Quantitative analysis revealed variations between different brands of MDMA tablets and within the same brand. In 1990–2009, the average MDMA content of UK tablets was ∼50–80 mg, range 20–131 mg, but 96% of tablets contained less than 100 mg MDMA per tablet and with a bimodal distribution of 20–40 mg and 60–80 mg MDMA per tablet, as reported by drug checking services and forensic institutes.32 53% of all ecstasy tablets tested in 2015 contained over 140 mg of MDMA compared to just 3% in 2009. In 2016, the average MDMA content was ∼125 mg MDMA per tablet.33 There are also “super pills” found on the market in some countries with a reported range of 270–340 mg. Worryingly, there are reports of large variations in the dosage in similar looking tablets.33 In this study, most of the doses quantified are significantly more than those earlier average doses of UK MDMA. Many tablets, especially tablets number 4 and 5, contain from double to more than double the dose of MDMA (160–165 mg) required to produce a physiological effect (70 mg or between 1–2 mg kg−1).7,341H q-NMR allowed not only the quantification of the dose of MDMA, but also facilitated both detection and quantification of any protonated impurities present in the tablets. Caffeine was detected and quantified in tablets 11a and b, containing 16.72 mg and 17.93 mg respectively with an RSD < 3%. Caffeine signals N-methyl 10, N-methyl 14 and aromatic 8 were used for quantification,35 while N-methyl 12 was not integrated due to its close proximity to the methine at position 2 of MDMA (Fig. 4). UHPLC using UV detection resulted in a broad peak at 4.7 min for caffeine, and the MS analysis showed a weak [M + H]+ at 195.0874 m/z, required for C8H11N4O2 195.0876 (Fig. 4).
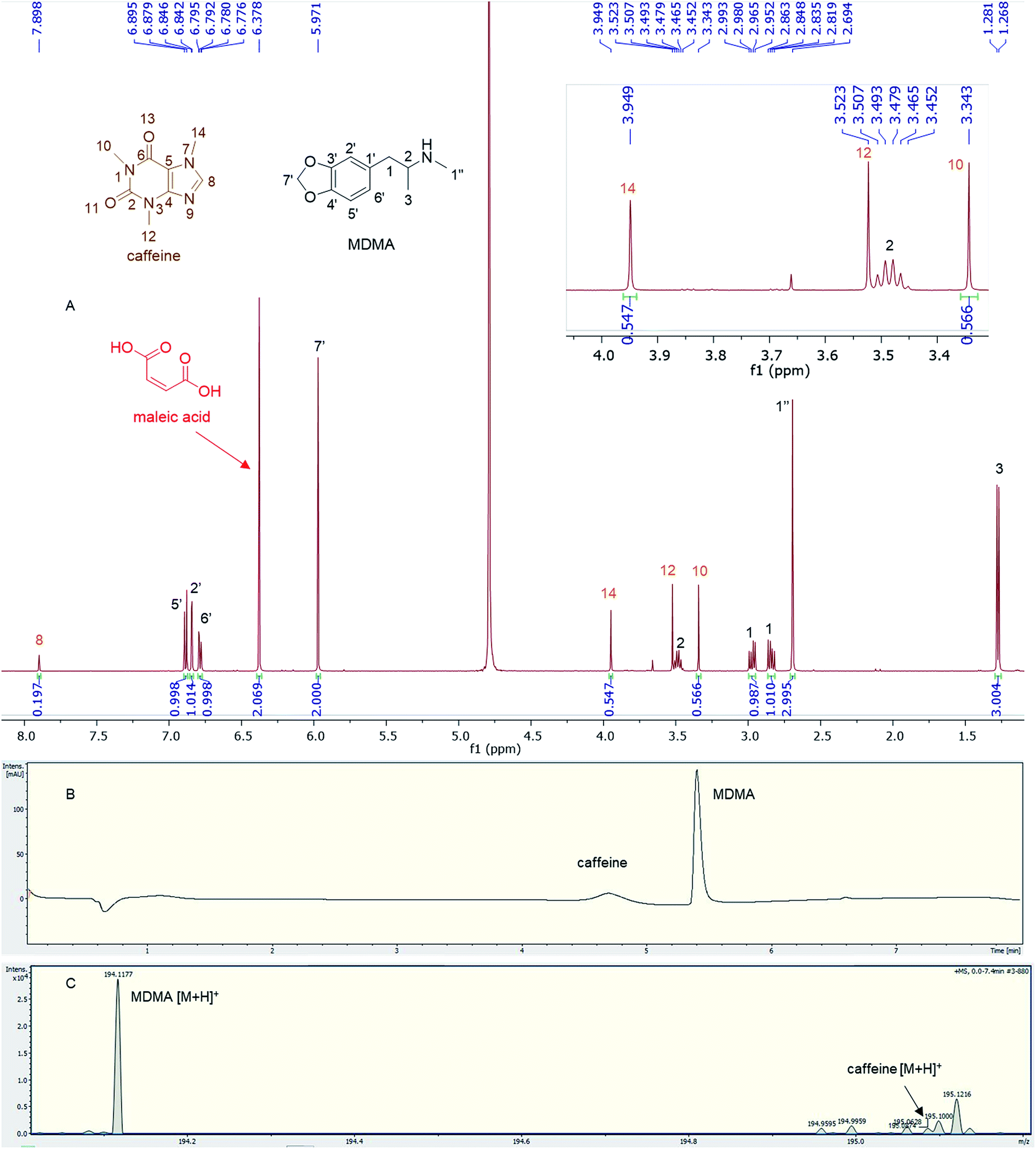 | ||
| Fig. 4 (A) 1H NMR (in D2O) of tablet 11a showing the caffeine peaks with expansion of the region between 3.4–4.5 ppm, (B) UHPLC of tablet 11a showing a broad peak for caffeine at RT = 4.7 min, (C) MS trace of tablet 11a showing MDMA molecular ion and caffeine low intensity molecular ion at 195.0874 m/z. | ||
Also shown are 1H NMR spectra (in D2O) of other seized MDMA samples cut or contaminated, e.g., with its dimethyl analogue, N,N-dimethyl-3,4-methylenedioxyamphetamine (MDDMA), reported to be found in MDMA samples synthesized through a nitropropene and reductive amination route.36 Due to the similar chemical structures, and therefore similar magnetic environments, the NMR spectra displayed multiple overlapping signals especially in the aromatic region and the methylene protons resonating at 2.73–3.10 ppm (Fig. 5). A total of 7 protons, 2 × N-CH3 plus one CH from the methylene overlapped with one of the MDMA methylene doublet of doublets at 2.78 ppm. Quantitatively, MDDMA comprised 10% of the sample.
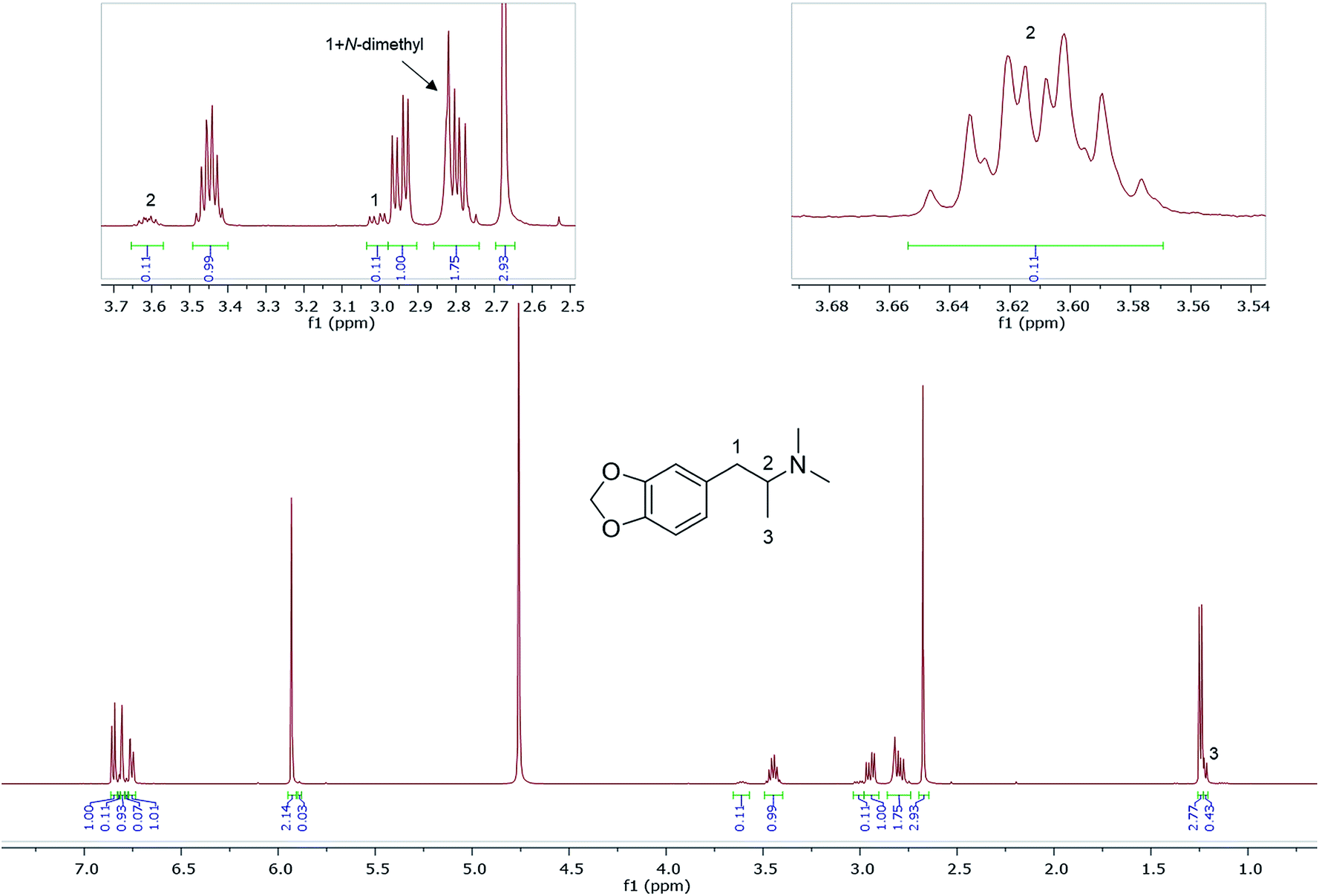 | ||
| Fig. 5 1H NMR (in D2O) of sample HN165, MDMA containing MDDMA impurity with expansions revealing signals for MDDMA positions 1, 2 and N-dimethyl. | ||
1H NMR data are reported for MDMA cut with 4-bromo-2,5-dimethoxyphenethylamine (2C-B) (Fig. 6) together with the assignment of the two p-methoxy functional groups using NOESY NMR data. 2C-B in combination with MDMA (83% 2C-B, 17% MDMA as molar ratios) was seized as a ground-up orange tablet supplied in a small plastic packet. First synthesized by Shulgin in the mid-1970s,37 the 2C-B pharmacological profile is similar to that of MDMA, it primarily inhibits 5-HT transporters. It also has less potency for dopamine and noradrenalin transporters. There has been a report of tablets sold as MDMA, but rather containing other psychoactive drugs: 3,4-methylenedioxyamphetamine (MDA), TFMPP, 2C-B, and caffeine.38
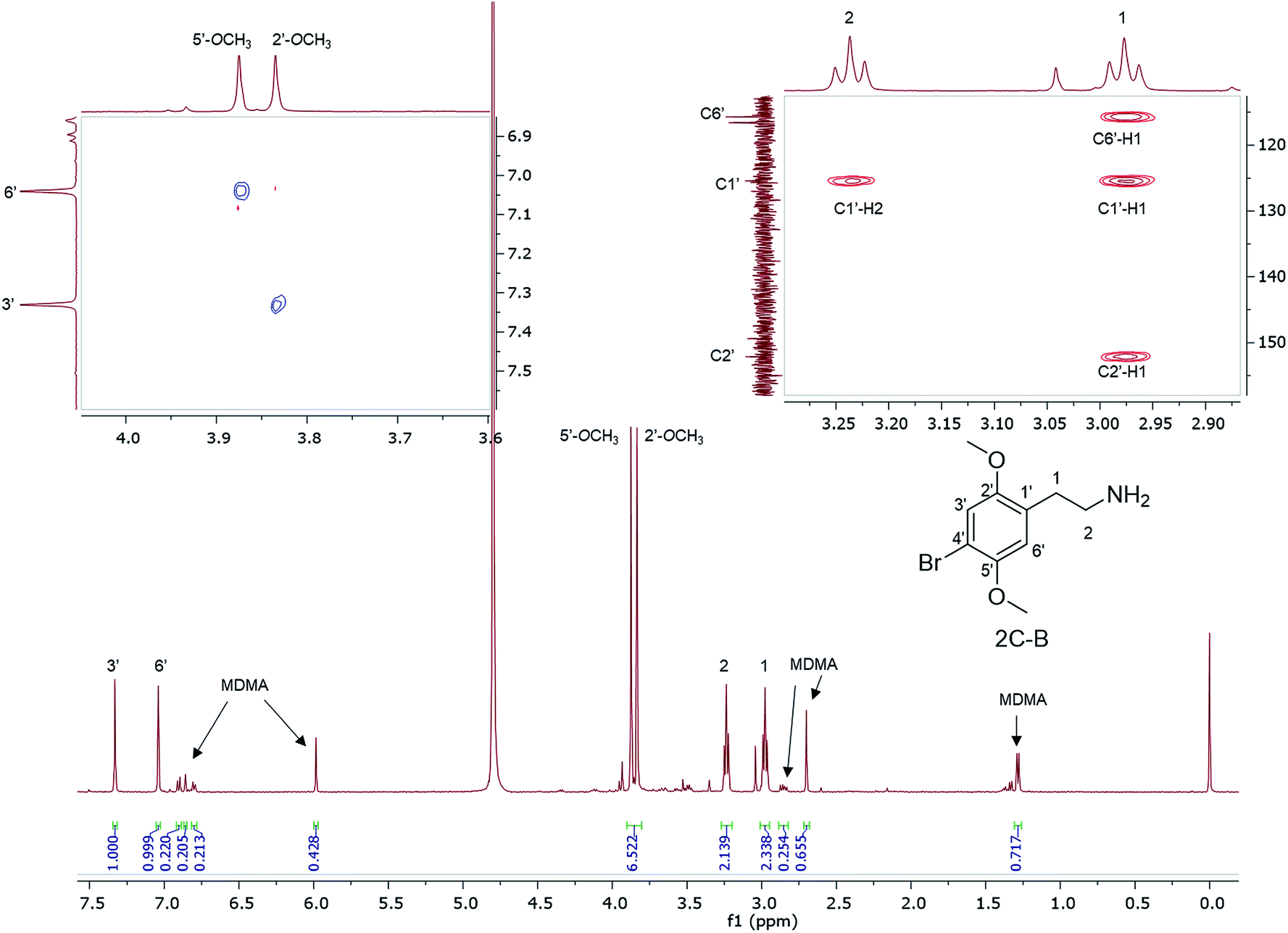 | ||
| Fig. 6 1H NMR (in D2O) of a 2C-B and MDMA mixture with TMSP 0.00 ppm, (top right) expansion of the HMBC spectra showing key HMBC connectivities, (top left) expansion of NOESY spectra revealing key NOE cross-peak connectivities. | ||
4-Iodo-2,5-dimethoxyphenethylamine (2C-I), the iodo analogue of the 2C family, was detected in sample HN144, a blue powder, in combination with 2C-B and MDMA with a molar percentage of 50% 2C-I, 41% 2C-B and 9% MDMA. The ESI-MS analysis revealed 2C-I [M + H]+ 308.0130 m/z for C10H15INO2 requires 308.0147. 2C-B [M + H]+ 260.0264 m/z for C10H1579BrNO2 requires 260.0286, and [M + H]+ 262.0253 m/z for C10H1581BrNO2 requires 262.0260 (ratio 1:1).39 The 1H NMR of 2C-I and 2C-B overlapped with the exception of the aromatic signals, where the presence of the iodo substituent at the p-aromatic position resulted in a greater chemical shift difference in the 1H spectra between the meta- (3′) (7.49 ppm) and the ortho- (6′) (6.96 ppm) protons compared with that found in 2C-B (Fig. 7). This difference is of diagnostic value in determining the type of substituent in the NPS 2C family by 1H NMR spectroscopy. A survey conducted for self-reporting NPS use on 682 attendees aged between 16–25 at electronic dance music festivals in New York (2015), found that 35% reported the use of NPS including cathinones, with methylone being the most popular cathinone and 2C-I the most popular phenethylamine NPS. The survey also highlighted the multidrug use of MDMA with other NPS and its risk factors for intoxication and possibly death.40 In Fig. 7, MDMA is shown to be only a minor component (9%) mixed with 2C-I and 2C-B, but still dangerously sold as “ecstasy”. This is an excellent example that “you do not know what you are buying”, if another one was needed.
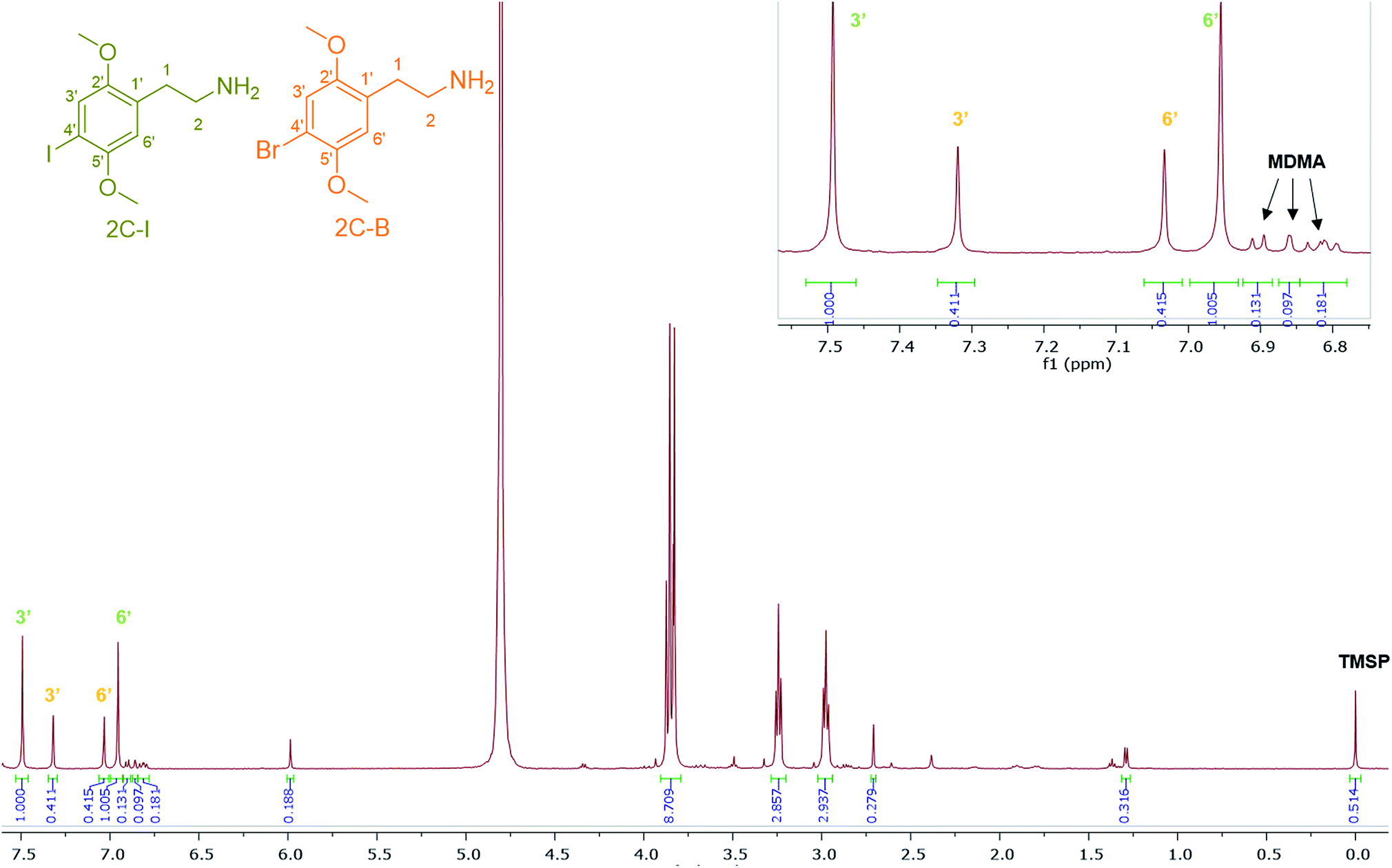 | ||
| Fig. 7 1H NMR (in D2O) spectra of sample HN144 containing 2C-I, 2C-B, and MDMA with expansion of the aromatic region between 6.80–7.55 ppm. | ||
Conclusions
In this paper, 1H q-NMR using the MA IS method was applied successfully to complex samples of MDMA cut with other NPS of closely similar structures seized from night-club venues. A cross-method validation of 1H q-NMR to analyse seized MDMA tablets is described. 1H q-NMR proved its versatility in the identification and quantification of the mixing/cutting agents as well as MDMA, with less sample handling (no serial dilution) and better precision (RSD%) compared to chromatographic and MS-based techniques. Comparative analysis with such chromatographic and MS-based methods further corroborated the results of 1H q-NMR which provided a fast (15 min), reproducible quantification without the use of a reference standard of the analyte under investigation. MA is a suitable NMR quantitative IS due to its high solubility, simplicity of the peak and non-overlapping signal. Identification of tablets containing such varying amounts of MDMA is a cause of concern to the public. It is also of value to law enforcement officials and health workers. When certain parameters are carefully optimized and considered such as the relaxation delay, number of scans, and S/N, NMR possesses the accuracy and reliability in the quantitative analysis of MDMA in seized tablets with results comparable to other gold standard analytical techniques, e.g. GC and LC.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We acknowledge the Drug Expert Action Team (DEAT), Avon and Somerset Constabulary, for the provision of seized samples, and the Government of Kuwait for a fully funded studentship (to HAN).References
- European Monitoring Centre for Drugs and Drug Addiction (EMCDDA), The European Drug Report 2018: Trends and Developments, 2018, http://www.emcdda.europa.eu/system/files/publications/8585/20181816_TDAT18001ENN_PDF.pdf, accessed 08/08/19 Search PubMed.
- European Monitoring Centre for Drugs and Drug Addiction (EMCDDA), The European Drug Report 2016: Trends and Developments, 2016, EMCDDA, Lisbon, http://www.emcdda.europa.eu/publications/edr/trends-developments/2016, accessed 08/08/19 Search PubMed.
- European Monitoring Centre for Drugs and Drug Addiction (EMCDDA), The European Drug Report 2019: Trends and Developments, 2019, EMCDDA, Lisbon, http://www.emcdda.europa.eu/publications/edr/trends-developments/2019_enpp48-50, accessed 08/08/19 Search PubMed.
- J. P. Kelly, Cathinone derivatives: a review of their chemistry, pharmacology and toxicology, Drug Test. Anal., 2011, 3, 439–453, DOI:10.1002/dta.313.
- L. J. De Felice, R. A. Glennon and S. S. Negus, Synthetic cathinones: chemical phylogeny, physiology, and neuropharmacology, Life Sci., 2014, 97, 20–26 CrossRef CAS PubMed.
- M. L. Banks, T. J. Worst, D. E. Rusyniak and J. E. Sprague, Synthetic cathinones (“bath salts”), J. Emerg. Med., 2014, 46, 632–642 CrossRef PubMed.
- P. Armenian, T. M. Mamantov, B. T. Tsutaoka, R. R. Gerona, E. F. Silman, A. H. Wu and K. R. Olson, Multiple MDMA (ecstasy) overdoses at a rave event: a case series, J. Intensive Care Med., 2012, 28, 252–258, DOI:10.1177/0885066612445982.
- D. Zuba and B. Byrska, Analysis of the prevalence and coexistence of synthetic cannabinoids in “herbal high” products in Poland, Forensic Toxicol., 2013, 31, 21–30 CrossRef CAS.
- A. L. A. Mohr, M. Friscia, J. K. Yeakel and B. K. Logan, Use of synthetic stimulants and hallucinogens in a cohort of electronic dance music festival attendees, Forensic Sci. Int., 2018, 282, 168–178, DOI:10.1016/j.forsciint.2017.11.017.
- D. Lee, C. W. Chronister, J. Hoyer and B. A. Goldberger, Ethylone-related deaths: toxicological findings, J. Anal. Toxicol., 2015, 39, 567–571, DOI:10.1093/jat/bkv053.
- J. P. Smith, O. B. Sutcliffe and C. E. Banks, An overview of recent developments in the analytical detection of New Psychoactive Substances (NPSs), Analyst, 2015, 140, 4932–4948, 10.1039/c5an00797f.
- L. R. Cumba, J. P. Smith, K. Y. Zuway, O. B. Sutcliffe, D. R. do Carmoa and C. E. Banks, Forensic electrochemistry: simultaneous voltammetric detection of MDMA and its fatal counterpart “Dr Death” (PMA), Anal. Methods, 2016, 8, 142–152, 10.1039/c5ay02924d.
- H. M. Elbardisy, C. W. Foster, L. Cumba, O. B. Sutcliffe and C. E. Banks, et al., Analytical determination of heroin, fentanyl and fentalogues using high-performance liquid chromatography with diode array and amperometric detection, Anal. Methods, 2019, 11, 1053–1063 RSC.
- H. M. Elbardisy, A. G. M. Ferrari, C. W. Foster, O. B. Sutcliffe, D. A. C. Brownson, T. S. Belal, W. Talaat, H. G. Daabees and C. E. Banks, Forensic electrochemistry: the electroanalytical sensing of mephedrone metabolites, ACS Omega, 2019, 4, 1947–1954 CrossRef CAS.
- R. C. Schneider and K. A. Kovar, Analysis of ecstasy tablets: comparison of reflectance and transmittance near infrared spectroscopy, Forensic Sci. Int., 2003, 134, 187–195 CrossRef CAS.
- P. A. Hays, Proton nuclear magnetic resonance spectroscopy (NMR) methods for determining the purity of reference drug standards and illicit forensic drug seizures, J. Forensic Sci., 2005, 50, 1342–1360 CrossRef CAS.
- G. Frison, M. Gregio, L. Zamengo, F. Zancanaro, S. Frasson and R. Sciarrone, Gas chromatography/mass spectrometry determination of mephedrone in drug seizures after derivatization with 2,2,2-trichloroethyl chloroformate, Rapid Commun. Mass Spectrom., 2011, 25, 387–390, DOI:10.1002/rcm.4842.
- F. Malz and H. Jancke, Validation of quantitative NMR, J. Pharm. Biomed. Anal., 2005, 38, 813–823 CrossRef CAS PubMed.
- U. Holzgrabe, Quantitative NMR spectroscopy in pharmaceutical applications, Prog. Nucl. Magn. Reson. Spectrosc., 2010, 57, 229–240 CrossRef CAS PubMed.
- N. S. Almeida, L. E. C. Benedito, A. O. Maldaner and A. L. de Oliveira, A validated NMR approach for MDMA quantification in ecstasy tablets, J. Braz. Chem. Soc., 2018, 29, 1944–1950, DOI:10.21577/0103-5053.20180071.
- L. H. Antonides, R. M. Brignall, A. Costello, O. B. Sutcliffe and R. E. Mewis, et al., Rapid identification of novel psychoactive and other controlled substances using low-field 1H NMR spectroscopy, ACS Omega, 2019, 4, 7103–7112 CrossRef CAS PubMed.
- G. Assemat, F. Dubocq, S. Balayssac, C. Lamoureux, M. Malet-Martino and V. Gilard, Screening of “spice” herbal mixtures: from high-field to low-field proton NMR, Forensic Sci. Int., 2017, 279, 88–95, DOI:10.1016/j.forsciint.2017.08.006.
- Y. Zhong, K. Huang, Q. Luo, S. Yao, X. Liu, N. Yang, C. Lin and X. Luo, The application of a desktop NMR spectrometer in drug analysis, Int. J. Anal. Chem., 2018, 2018, 3104569, DOI:10.1155/2018/3104569.
- J. Duffy, A. Urbas, M. Niemitz, K. Lippa and I. Marginean, Differentiation of fentanyl analogues by low-field NMR spectroscopy, Anal. Chim. Acta, 2019, 1049, 161–169, DOI:10.1016/j.aca.2018.12.014.
- European Network of Forensic Science Institutes – Drugs Working Group, Guidelines on Sampling of Illicit Drugs for Quantitative Analysis, 2014, http://enfsi.eu/wp-content/uploads/2016/09/guidelines_quant_sampling_dwg_printing_vf4.pdf, accessed 04/09/19 Search PubMed.
- G. Maniara, K. Rajamoorthi, S. Rajan and G. W. Stockton, Method performance and validation for quantitative analysis by 1H and 31P NMR spectroscopy. Applications to analytical standards and agricultural chemicals, Anal. Chem., 1998, 70, 4921–4928 CrossRef CAS PubMed.
- H. H. Gadape and K. S. Parikh, Quantitative determination and validation of Metformin hydrochloride in pharmaceutical using quantitative nuclear magnetic resonance spectroscopy, E-Journal of Chemistry, 2011, 8, 767–781, DOI:10.1155/2011/461898.
- I. B. Müller and C. N. Windberg, Validation of an HPLC method for quantitation of MDMA in tablets, J. Chromatogr. Sci., 2005, 43, 434–437 Search PubMed.
- S. Wang, M. Cyronak and E. Yang, Does a stable isotopically labeled internal standard always correct analyte response? A matrix effect study on a LC/MS/MS method for the determination of carvedilol enantiomers in human plasma, J. Pharm. Biomed. Anal., 2007, 43, 701–707 CrossRef CAS PubMed.
- A. S. Davison, A. M. Milan and J. J. Dutton, Potential problems with using deuterated internal standards for liquid chromatography-tandem mass spectrometry, Ann. Clin. Biochem., 2013, 50, 274 CrossRef CAS PubMed.
- Sigma-Aldrich, Quantitative NMR: Technical details and TraceCERT Certified Reference Materials, 2017, https://www.sigmaaldrich.com/content/dam/sigma-aldrich/docs/Sigma-Aldrich/Brochure/1/qnmr-brochure-rjo.pdf, accessed 03/07/19 Search PubMed.
- D. M. Wood, V. Stribley, P. I. Dargan and J. Ramsey, et al., Variability in the 3,4-methylenedioxy-methamphetamine content of ‘ecstasy’ tablets in the UK, Emerg. Med. J., 2011, 28, 764–765 CrossRef PubMed.
- European Monitoring Centre for Drugs and Drug Addiction (EMCDDA), Recent changes in Europe's MDMA/ecstasy market, 2016, http://www.emcdda.europa.eu/system/files/publications/2473/TD0116348ENN.pdf, accessed 03/07/19 Search PubMed.
- European Monitoring Centre for Drugs and Drug Addiction (EMCDDA), Methylenedioxymethamphetamine (MDMA or ‘Ecstasy’) drug profile, 2019, http://www.emcdda.europa.eu/publications/drug-profiles/mdma, accessed 03/07/19 Search PubMed.
- J. Sitkowski, L. Stefaniak, L. Nicol, M. L. Martin, G. J. Martin and G. A. Webb, Complete assignments of the 1H, 13C and 15N NMR spectra of caffeine, Spectrochim. Acta, 1995, 51, 839–841, DOI:10.1016/0584-8539(94)00192-e.
- P. Gimeno, F. Besacier, M. Bottex, L. Dujourdy and H. Chaudron-Thozet, A study of impurities in intermediates and 3,4-methylenedioxymethamphetamine (MDMA) samples produced via reductive amination routes, Forensic Sci. Int., 2005, 155, 141–157 CrossRef CAS PubMed.
- A. Shulgin and A. Shulgin, PIHKAL: A Chemical Love Story, Transform Press, Berkeley, California, 1991 Search PubMed.
- L. R. Togni, R. Lanaro, R. R. Resende and J. L. Costa, The variability of ecstasy tablets composition in Brazil, J. Forensic Sci., 2014, 60, 147–151 CrossRef PubMed.
- C. Giroud, M. Augsburger, L. Rivier, P. Mangin, F. Sadeghipour, E. Varesio, J. L. Veuthey and P. Kamalaprija, 2C-B: a new psychoactive phenylethylamine recently discovered in ecstasy tablets sold on the Swiss black market, J. Anal. Toxicol., 1998, 22, 345–354 CrossRef CAS PubMed.
- J. J. Palamar, P. Acosta, S. Sherman, D. C. Ompad and C. M. Cleland, Self-reported use of novel psychoactive substances among attendees of electronic dance music venues, Am. J. Drug Alcohol Abuse, 2016, 42, 624–632 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ay01403a |
| This journal is © The Royal Society of Chemistry 2019 |