
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReview of 3D cell culture with analysis in microfluidic systems
Andre D.
Castiaux
a,
Dana M.
Spence
b and
R. Scott
Martin
 *a
*a
aDepartment of Chemistry, Saint Louis University, 3501 Laclede Ave. St. Louis, MO 63103, USA. E-mail: scott.martin@slu.edu
bDepartment of Biomedical Engineering, Institute for Quantitative Health Science & Engineering, Michigan State University, East Lansing, MI 48824, USA. E-mail: spenceda@msu.edu
First published on 6th August 2019
Abstract
A review with 105 references that analyzes the emerging research area of 3D cell culture in microfluidic platforms with integrated detection schemes. Over the last several decades a central focus of cell culture has been the development of better in vivo mimics. This has led to the evolution from planar cell culture to cell culture on 3D scaffolds, and the incorporation of cell scaffolds into microfluidic devices. Specifically, this review explores the incorporation of suspension culture, hydrogels scaffolds, paper-based scaffolds, and fiber-based scaffolds into microfluidic platforms. In order to decrease analysis time, simplify sample preparation, monitor key signaling pathways involved in cell-to-cell communication or cell growth, and combat the limitations of sample volume/dilution seen in traditional assays, researchers have also started to focus on integrating detection schemes into the cell culture devices. This review will highlight the work that has been performed towards combining these techniques and will discuss potential future directions. It is clear that microfluidic-based 3D cell culture coupled with quantitative analysis can greatly improve our ability to mimic and understand in vivo systems.
Introduction
Cell culture, as it is known today, began in the late 1800's with the harvesting of cells from animal tissue and culturing or maintaining the tissue in natural media such as lymph fluid or plasma.1–3 As interest in different cell lines grew, an understanding of what was necessary for cell health and longevity also grew. This spurred the development of partially synthetic, and then fully synthetic media for cell culture (1900's – present).4,5 This has resulted in the development of over 3600 available cell lines originating from over 150 different species according to the American Type Culture Collection.6 Using these widely available cell types, researchers have been able to expand our understanding of formation and function of tissues in the body, which has ultimately led to great advances in medicine and health.The most popular method for maintaining and analyzing adherent cell lines over the last century has been 2D or monolayer culture.7,8 In 2D culture, cells are maintained on a flat surface, most often a plastic culture flask or Petri dish (Fig. 1A). These surfaces can be easily modified with proteins or other surface functionalities to aid in cell adhesion and increase cell health.9–11 The cost of maintaining cells in these flasks is low and protocols have been developed that are robust and simple, reducing the likelihood of contamination. One can image directly through these substrates making it easy to monitor cell viability and morphology. It is also easy to add reagents and aliquot out samples to analyze the effect of various conditions and stimulations. However, in recent years it has become abundantly clear that while 2D culture is useful, it does not always accurately reflect the 3D environment that cells experience in vivo.7,12
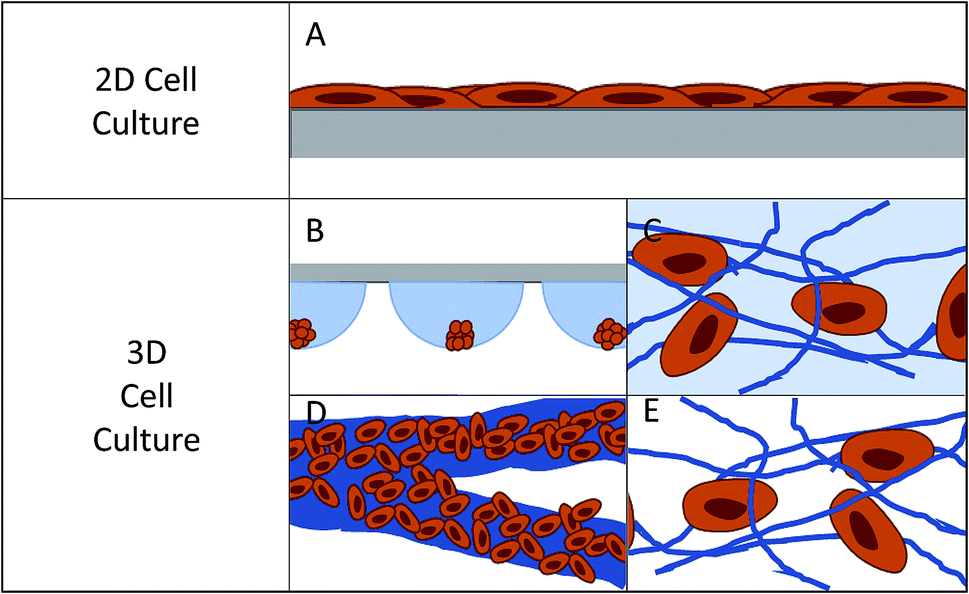 | ||
| Fig. 1 Pictorial representation of cells on 2D and 3D substrates. (A) Represents how cells lay on a planar substrate, where cells are polarized on one side and media contacts half of the cell surface. (B) Hanging drop cell culture: cells grow in a cluster where only the outer layers of cells have good contact to the culture medium. (C) Hydrogel cell culture: cells make discrete connections to hydrogel scaffold. The scaffold can be fibrous or more amorphous, with the scaffold being hydrated. (D) Paper-based cell culture: fibrous scaffold material where the fibers are typically much larger than the size of a cell. (E) Fiber-based cell culture: fibers can be tuned to sizes ranging from 10's of nanometers to 10's of microns. | ||
In 2D cell culture, the cells are limited to the surface of a single material, most often plastic (usually polystyrene). This plastic has defined surface characteristics such as roughness, stiffness, and limited binding sites. Due to the fact that the cell is forced to adhere within the plane of the surface, the area of the cell that is exposed to the bulk solution is limited (Fig. 1A).12 This causes several issues with the function of the cell. First, it limits the cell's exposure to chemical gradients by allowing solution access to only a fraction of the cell.7 Second, it can lead to a polarity shift within the cell, as the cell is forced to accommodate the new confined geometry, effecting cell function.7,13 Third, the cell is exposed to a continuous source of the surface matrix, in contrast to in vivo where cells have discrete attachments.12 Finally, the surface roughness is very low and the cells can proliferate and migrate without restraint, which is not an accurate mimic of in vivo conditions for many cell lines.12
In the body, cells interact with a complex extracellular matrix (ECM), where the complications listed above are optimized based on cell type and location within the body. In an attempt to develop systems that better mimic in vivo conditions, researchers are increasingly looking to 3D cell culture.14–16 A 3D cell culture platform allows cells to grow omni-directionally, unlike 2D cell culture that limits cells to a planar surface (Fig. 1).12 Naturally, a cell has a finite number of adhesion sites distributed in all three dimensions. 3D culture allows the cell to adopt a preferred orientation to optimize the cell polarity, and the cell polarity can change in reaction to a stimulus.12 Cell proliferation and migration are hindered due to the complex network of fibrils, but signaling molecules and chemical gradients are not hindered and are accessible to the entirety of the cell. It has repeatedly been shown that taking into account the ECM has beneficial effects on the tissue.15,17–19 3D cell cultures have shown differences in drug response, morphology, and proliferation rates under static culture conditions,7,20–23 while under flow conditions, response and function is enhanced to a greater extent.24 One way to introduce flow to cell culture systems is through the use of microfluidic devices.
Microfluidic systems can be produced in a variety of ways. Polydimethylsiloxane (PDMS) is the preferred material for making microfluidic devices due to its long history, ease of use, gas permeability, and relative low cost.25,26 However, these devices experience problems with lab-to-lab reproducibility and can be time consuming to make. PDMS is also gas permeable, which has advantages for oxygen diffusion into cell culture media but presents problems for long term cell culture where media evaporation can lead to osmolality shifts and possible complications with cell culture.26,27 Additionally, the uncured monomer can leach into the cell culture media or small molecules can diffuse into the material making analyte analysis difficult.26,28 To combat the complications presented by PDMS, microfluidic devices have also been made out of thermo plastics and glass, although each presents challenges in fabrication.29 More recent (last 10 years) advancements have been made in additive manufacturing, greatly reducing the time of fabrication and increasing lab-to-lab reproducibility. Additive manufacturing, or 3D printing technology, involves automated device assembly in a layer by layer fashion. Extrusion-based printing, stereolithography, and Polyjet are three main techniques of 3D printing applicable to microfluidics.30–32 Each of the above techniques can produce devices with channels that reach physiologically relevant dimensions.30,33 This is important for blood cells or endothelial cells that experience shear induced stress in vivo. This can allow more mimetic cell-to-cell communication.
Microfluidic techniques, in conjunction with cell culture, will open avenues for direct analysis of important signaling molecules. Optically transparent materials such as PDMS, glass, or some 3D printed plastics enable direct imaging of fluorescently tagged proteins and cells.25,28,34–36 In addition, more complex UV-VIS absorbance or fluorescent quantification can be performed for small molecules.24,33,37 Recently, methods for coupling mass spectrometers and electrochemical detectors to microfluidic systems have been developed.38–40 These detection schemes have been facilitated by separation and isolation techniques that are well established on microfluidic chips, including microchip electrophoresis and solid phase extraction.40–42
The integration of conventional 2D cell cultures into microfluidic devices is well characterized and common. However, the benefits of 3D cell culture vastly outweigh the ease in which 2D cell culture can be integrated into microfluidic platforms. Recently, work has shifted to incorporate 3D matrixes into microfluidic platforms, merging the advantages of small volumes and minimal dilution of microfluidics with the in vivo behavior of 3D cell culture. This review will discuss the current techniques that have been used to incorporate 3D cell culture into microfluidic platforms. This review will highlight the benefits of each 3D cell culture technique as it relates to cell function, and the ability to develop microfluidic systems to monitor complex cell-to-cell interactions with an emphasis on small molecule detection and quantitation as part of the device function. Ultimately the goal of this review is to provide background on current approaches to perform 3D cell culture in microfluidic devices with a focus on the various methods used to detect key analytes involved in cell-to-cell communication or cell growth.
Methods of 3D cell culture
Currently there are several different methods for 3D cell culture, with each method having benefits and disadvantages. In general, the techniques can be split into four categories: suspension (Fig. 1B), hydrogels (Fig. 1C), paper-based culture (Fig. 1D), and fiber scaffolds (Fig. 1E). Suspension techniques involve mostly free-floating clusters of cells, while the last three require a network on which the cells can adhere and proliferate.Suspension
The general class of suspension cell culture is the oldest form of 3D cell culture and has been around since the development of cell culture.2 However, suspension cell culture has been limited to mostly insect cell lines or mammalian cell lines that can be altered to grow under suspension conditions (non-adherent cells).43,44 This is because most mammalian cell lines are adherent cells that require a surface or scaffold to support their growth. The simplest method of suspension cell culture requires a non-treated culture flask that is agitated to keep cells suspended while they proliferate. Large scale suspension cell culture has primarily been used for the expression of proteins for use as therapeutics or in other bio-reactor applications.43–45A sub technique of suspension cell culture, which was developed in the late 1800's, is hanging drop cell culture.2 This technique relies on a suspended droplet hanging from a scaffold (Fig. 1B). Within this droplet, a single cell can be cultured into a cluster. While primarily used in embryology, this technique has found applications with a wide variety of mammalian cell lines.46–48 This technique is time consuming, often requiring many repetitive pipetting steps that greatly decrease throughput.
Coupling microfluidics with hanging drop cell culture methods can overcome some of these limitations. Wu et al. developed a PDMS based device, shown in Fig. 2A, to pump solution through the hanging drops based on pressure differences in reservoirs connected to the microfluidic chip.49 In this application, the researchers were able to easily introduce the cells into the droplets and then continuously flow fresh media through the droplets (Fig. 2B). Using this device, embryoid bodies were cultured in a high-throughput manner without the tedium of pipetting to supply fresh media.49
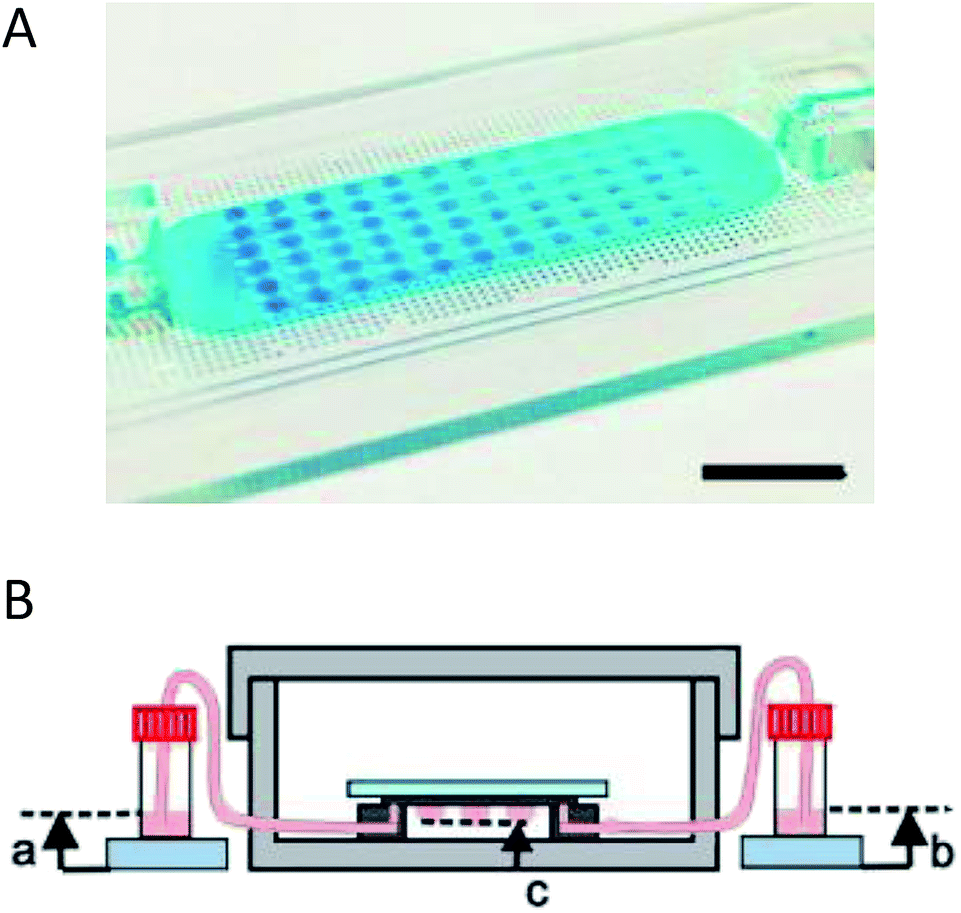 | ||
| Fig. 2 Microfluidic hanging drop cell culture chip. (A) Picture of a PDMS based microfluidic chip for hanging drop cell culture. Scale bar is 1 cm. (B) Illustration of the hanging drop microfluidic cell culture chip attached to reservoirs to supply fresh media to the cells. The chip is placed in a 10 cm dish containing PBS to minimize evaporation of the droplets. (Adapted from ref. 49 with permission from MDPI). | ||
More complex examples developed by Frey et al. incorporated valves and mixing channels into a 2D array of interconnected wells (Fig. 3A).50 This device allowed selective introduction of cells down each column via the cell loading ports (Fig. 3B) followed by flow perpendicular to the loading direction to allow for cell-to-cell communication (Fig. 3B). Using this device, the researchers were able to reproducibly culture spherical microtissues of different cell types for more than 10 days, with no significant difference between cell function using this technique and static spheroid culture. However, the microfluidic technique did not require the labor and time intensive pipetting steps traditionally used.50
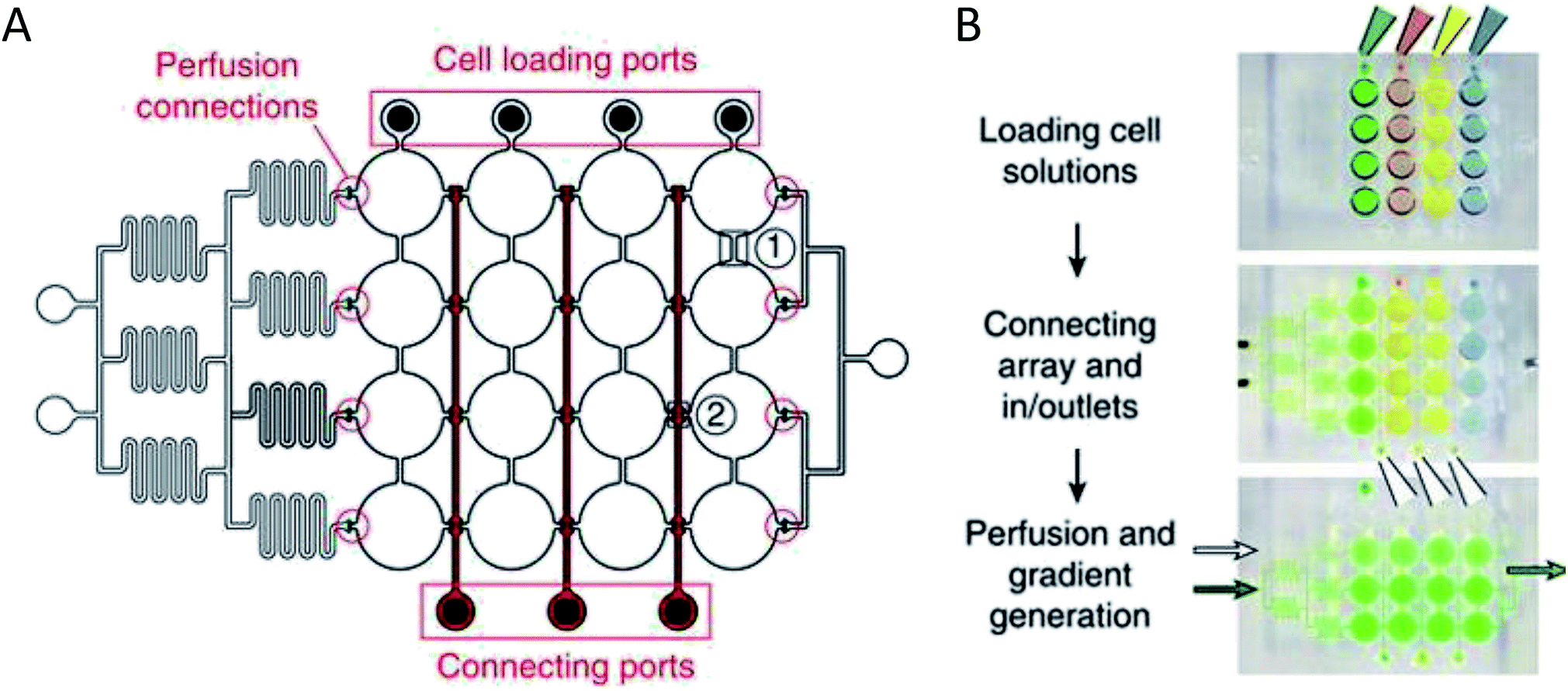 | ||
| Fig. 3 PDMS base hanging drop cell culture chip with valving. (A) Schematic representation of the PDMS based hanging drop cell culture chip. The chip has valves between each column allowing selective loading of cells down the channels. The valves can then be opened to allow flow from separate perfusion channels. (B) Schematic showing how the chip is utilized. First cells are loaded from the top down the channels. The valves are opened and flow is initiated from the perfusion reservoirs (left to the right). Buffer can be mixed with a desired compound and diluted inline to the wells, allowing a concentration gradient to develop down the wells. (Adapted from ref. 50 with permission from Springer Nature). | ||
Hydrogels
Most mammalian cells in vivo proliferate on a complex scaffold known as the extra cellular matrix (ECM). In the body this is most often in the form of a hydrated protein matrix that allows cell adhesion as well as a template for 3D cell growth. One solution to better mimicking this system was the development of hydrogels for cell culture.51,52 Hydrogels are composed of one or many different hydrophilic polymers having a polymerization pattern that enables cells and molecules to move in and out of the pores (Fig. 1B). These hydrogel systems provide a 3D scaffold for cell growth and allow small molecules access to more of the cell than planar substrates.Hydrogels can be made from a wide range of synthetic (polyacrylamide, polyethylene glycol, poly(acrylic acid)) and natural polymers (collagen, fibrin, hyaluronic acid).19,51–54 This range of available materials allows fine tuning of properties important to cell health and function. These properties include porosity, stiffness, and degradation of the matrix. However, fine tuning these parameters can be time consuming and lead to hydrogel systems that are complicated to produce and keep stable.19,54 Hydrogels have thermal and mechanical limitations that limit the working conditions and the longevity of the material. It is possible to produce hydrogels on a large scale, but there can be issues in uniformity and batch-to-batch consistency.19,51 Cells can be introduced to hydrogel matrixes either during or after polymerization depending on the cell line used or experimental factors being explored.51
The seminal work incorporating hydrogels into microfluidic devices was performed by Beebe's group and utilized passive pumping in microchannel arrays (Fig. 4).55–57 Passive pumping moves solutions based on pressure differences. By placing a fresh droplet of media on the inlet reservoir, it will push fresh solution through the microchannel to the outlet reservoir until the pressure of droplet remaining at the inlet equals that formed at the outlet.56 This work utilized 3D collagen gels as the substrate for the co-culture of human breast carcinoma cells (T47D) and normal human mammary fibroblasts.58 A direct comparison between cells cultured in the microwell arrays and cells cultured in a standard 12-well culture plate was performed. Through image analysis of the cells, the group was able to determine that the cells could be cultured in microfluidic channels (2 μL) without effecting cell behavior.58
 | ||
| Fig. 4 Diagram showing the mechanism of passive pumping. Passive pumping depends on the radius of each droplet. The pressure from a small droplet of radius Ri will be larger than the pressure from a large droplet of radius Ro based on Laplace's law. (Adapted from ref. 57 with permission from The Royal Society of Chemistry). | ||
Beebe's original studies involving collagen gels proved viable for cell culture in a system that is also amenable to high throughput. Since the development in the late 2000's, the complexity of such systems has grown greatly. There are now techniques for obtaining aligned collagen gels within microchannels a video publication by Burkel et al.59 A more complex PDMS device was reported by Chung et al. in which a three channel system separated by a porous collagen gel was employed to observe the migration of endothelial cells under various conditions, including co-culture (Fig. 5).60 By culturing the endothelial cells in the center channel, they were able to use one side channel as a control and introduce growth factors or other cells into the remaining side channel. Endothelial cell health and migration was monitored by imaging on the PDMS-based device for 5 days.60
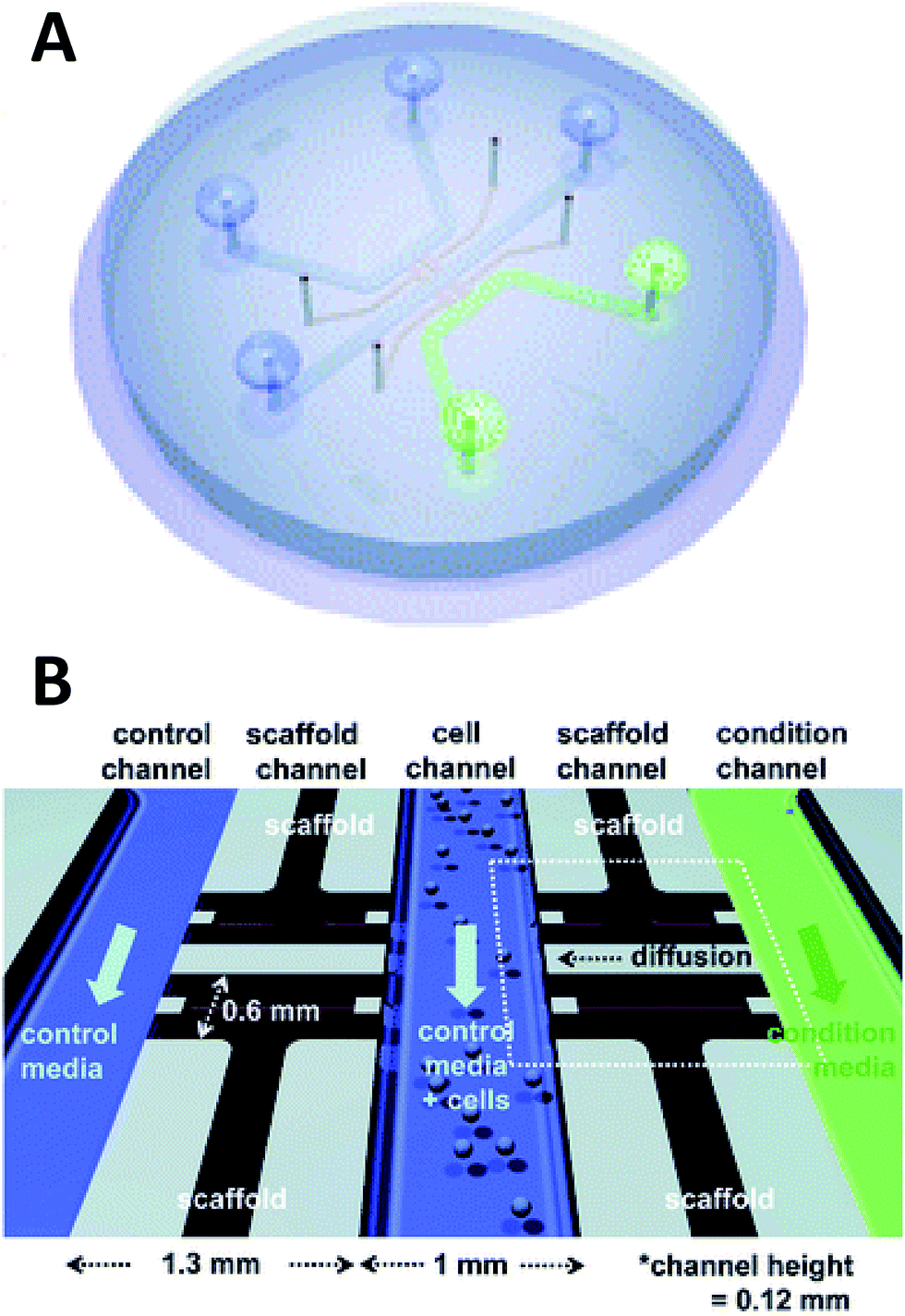 | ||
| Fig. 5 Microfluidic PDMS based platform for hydrogel based cell culture. (A) Completed chip with three distinct fluidic channels connected by hydrogel structures in between the channels. (B) Pictorial representation of the channel structure with flow channels in blue and green, and hydrogel structures in black. Cells are loaded in the center channel and cell migration is monitored in the gel structure based on stimulant in the side channels. (Adapted from ref. 60 with permission from The Royal Society of Chemistry). | ||
Another example of co-culture comes from Bischel et al. in which they explored the interplay between osteotropic prostate cancer cells and bone marrow stromal cells cultured in a hydrogel scaffold.61 Bone marrow stromal cells were cultured to the side of a microfluidic channel, followed by filling of the channel with prostate cancer cells embedded in collagen hydrogel (Fig. 6).61 Through imaging analysis, it was determined that co-culture of the osteotropic prostate cancer cells with the bone marrow stromal cells increased the invasive capacity of the cancer cells.61
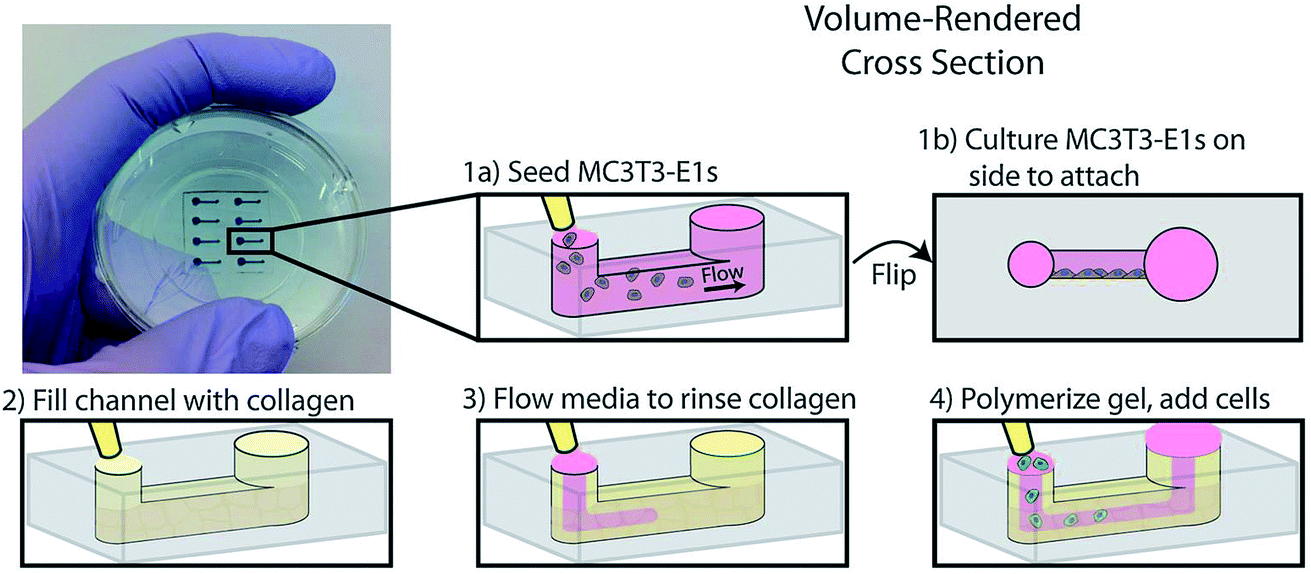 | ||
| Fig. 6 PDMS-based microfluidic co-culture hydrogel platform. MC3T3-E1 cells are seeded in the channel and the device is flipped 90° such that the cells adhere to the sidewall of the channel. The channel is then filled with a collagen solution, after which the collagen solution is replaced by culture media leaving a thin layer of collagen on the walls. This collagen is polymerized into a hydrogel and prostate cancer cells are seeded in the channel. (Adapted from ref. 61 with permission from Oxford University Press). | ||
More recently, 3D printing has become prevalent in microfluidic fabrication. Concurrent with this technology has been the development of 3D printed hydrogels. These new hydrogel fabrication techniques include stereolithography methods and extrusion-based bio-plotter methods.35,62–65 For now, most of the work has centered around the production of stand-alone hydrogel systems. However, through multi-material printing capabilities, 3D printed hydrogel scaffolds could be directly incorporated into rigid structures during the print process.
Paper cell culture
While hydrogels require the synthesis of material as needed, paper-based cell culture provides similar scaffolding that is less labor intensive. Paper-based cell culture relies on commercially available paper scaffolds for 3D cell culture.66–69 The cellulose matrix of paper allows a rigid scaffold that can be folded into complex geometries, as well as a porous structure for cell growth in a 3D format.66–69 The porous structure also serves as a mimic to monitor cell migration. Paper-based culture has distinct advantages over hydrogel applications when considering the material's rigidity and thermal stability,67 which enable a wide range of surface modifications including chemical treatment, plasma treatments, and sterilization techniques. Additionally, well-established patterning techniques utilizing hydrophobic wax allows defined microfluidic networks to be developed on a single sheet of paper or between multiple, stacked paper sheets.70–73 However, paper-based techniques do have limitations when considered as an in vivo mimic. The material is mostly cellulose-based and requires modification of the paper material with ECM proteins.67 Additionally, the fiber diameter for most paper-based systems is relatively large (>1 μm),74 compared to the ∼500 nm size of most fibrils in the body.75Incorporation of paper based scaffolds into microfluidics using traditional lithography techniques was reported by Martinez et al. in 2007.73 By the year 2009 the technique was independently revamped by Lu et al. and Carrilho et al. for high throughput production using wax printing technology.70,76 The use of wax printing to create hydrophobic barriers into the paper microfluidic devices has grown in complexity and includes the addition of cell culture.35,69,71,72 There are very few cases of direct cell culture on paper based microfluidic devices, although one such study by Tao et al. described a cell culture platform with four areas for cell culture connected by two channels in a T-design (Fig. 7).74 Cells were seeded directly on the culture areas by pipetting and behaved the same as cells cultured in standard hydrogel conditions. The use of paper in microfluidic devices is almost exclusively coupled with hydrogel culture. Some of the earliest applications used stacked paper to construct layered systems that were accessible to the cells. The layers are fluidically connected via hydrogel systems impregnated with cells in the paper scaffold (Fig. 8).66 Derda et al. was one of the first to report this method using MDA-MB-231 cells grown in 8 layers of paper.66 These layers, 200 μm thick each, represent the distance the cells are from fresh media. Using the paper layers to separate cells from fresh media requires oxygen, fresh nutrients, and cells secretions to diffuse in and out of the system through the microstructure developed by the paper.66 Ultimately the group was able to separate the layers to analyze cell proliferation as a function of depth in the system. Since the cells were still intact after separating, they were also able to characterize cell recovery after exposure to optimal growth conditions. Mosadegh et al. later developed a modified version to explore chemotaxis of cancer cells and discovered that different cell types could be classified by their rate of chemotaxis.77
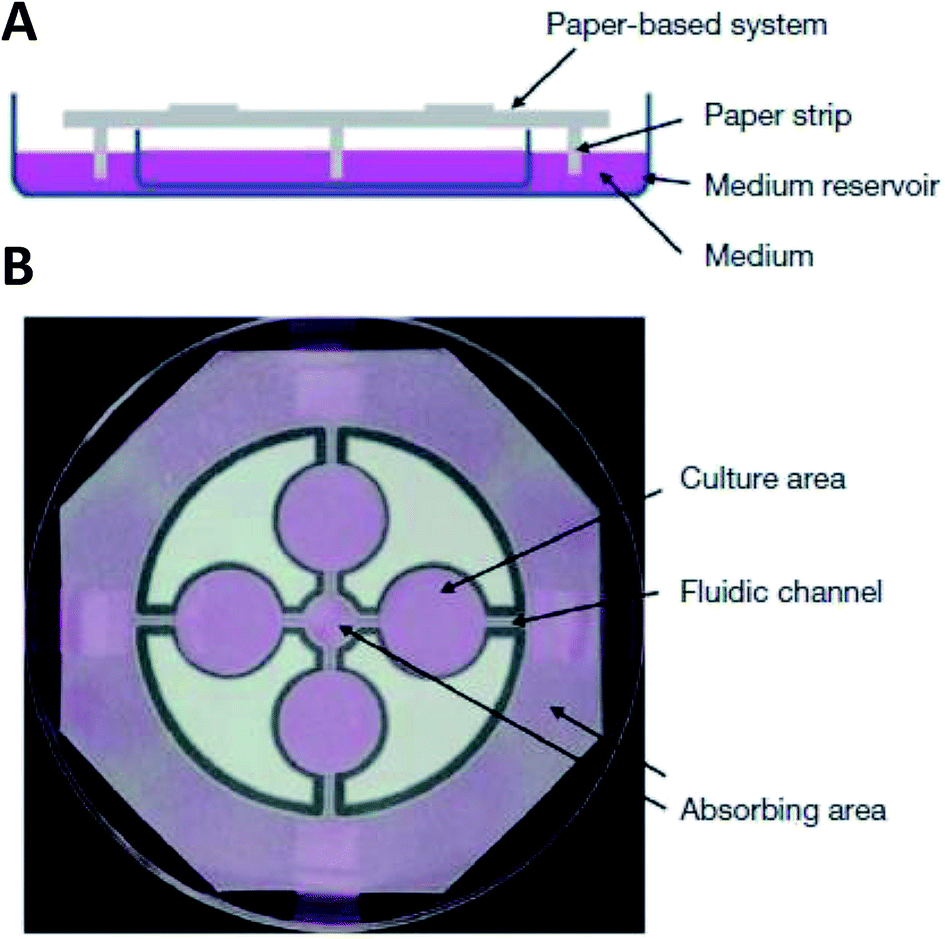 | ||
| Fig. 7 Paper-based microfluidic cell culture system. (A) The paper chip is suspended above a dish filled with medium. Strips of paper at the end of the chip allow connection to the medium reservoir. (B) Wax patterning defines 4 cell culture areas and channels to each cell culture area. (Adapted from ref. 74 with permission from Springer Nature). | ||
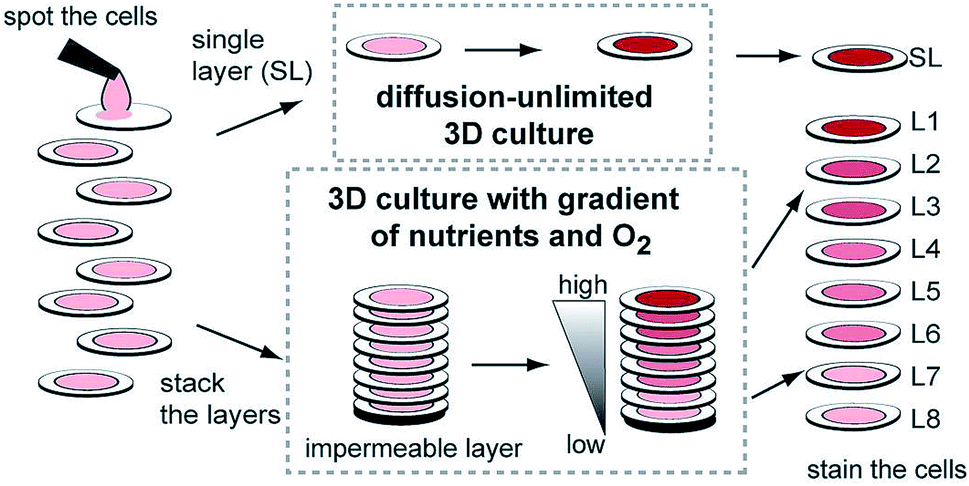 | ||
| Fig. 8 Stacked paper based 3D culture platform. Cells are spotted on paper (2–8 mm radius spots). The layers can then either be stacked to mimic a nutrient system (limited by diffusion) or left free to mimic a diffusion-unlimited 3D cell culture system. The stacked system can be disassembled for analysis. (Adapted from ref. 66 with permission from the Proceedings of the National Academy of Science). | ||
While the above work relied on the microfluidic structures of the paper, there are more complex examples in which microfluidic structures were patterned on paper substrate. Kenney et al. patterned channels onto Whatman 105 lens paper,78 which was then sandwiched between either a PDMS layer and a cellulose acetate layer or between two cellulose acetate layers. This format allowed the researchers to selectively control gas diffusion to the cell culture. Using this device, the chemotaxis of breast adenocarcinoma cells was monitored under different oxygen gradients and it was determined that these cells migrate based on available oxygen gradients.78 While the optical transparency of the device enabled imaging of the cells, later iterations incorporated a pH-sensing thin film directly under the cell culture scaffold.79 The thin film contained pH sensitive fluorescent dyes that could be imaged through the acrylic holder allowing real-time monitoring of the pH.79
Fiber scaffolds
The final 3D cell culture method discussed in this review involves the use of fiber scaffolds. It combines the freedom of material provided by hydrogels with the scaffold of paper-based cell culture. There are several methods for producing nanofibers that include solution blow spinning, melt spinning, and electrospinning.80–85 Each of these techniques can produce both natural (collagen, gelatin, silk fibroin) and synthetic fibers (polycaprolactone, polyurethane, polystyrene). The fibers can be tuned to sizes ranging from 10's of microns down to 10 nanometer diameters similar to the extracellular matrix.81,84 Various materials can be blended together to tune the structural properties of the material making the fiber mat stronger or preventing degradation.Fiber scaffolds have found a wide range of applications and can be used under static cell culture or under flow conditions. They are stable over a wide range of temperatures.80,86 Synthetic fibers are more stable in solution over long periods of time whereas natural fibers tend to degrade rapidly.80,86,87 Fiber scaffolds have biological compatibility to the point of being used in wound healing applications.17,88,89 However, production of electro-spun fibers often requires toxic volatile organic solvents, which have the potential to leach into cell cultures.90 Benign aqueous based solvents have been explored in limited applications and has been shown to minimize effects on protein structure.91,92
Early incorporation of fiber scaffolds into microfluidic devices was done with PDMS-based substrates. One of the earliest was reported in 2008 by Lee et al., who fabricated a channel in a bottom layer of PDMS and then used a top layer of PDMS to sandwich acrylic acid modified polyurethane fibers into the device (Fig. 9).93 A loading port open in the top layer enabled cells seeding from the top and culture. Using this device, the importance of the functionality of the extracellular matrix was demonstrated as human mesenchymal stem cell proliferation was faster on acrylic acid modified polyurethane nanofibers than on standard fibers.93 Similarly, Wallin et al. developed an array device in which they were able to pattern different fiber pads within a single microfluidic channel giving them control over parameters such as fiber diameter, alignment, or type (Fig. 10).94 This fiber array was then coupled to a microfluidic mixer to control drug or media gradients flowing over the cells cultured on the fiber pads. Neural stem cells could be imaged on aligned fibers, while simultaneously applying a chemical gradient to determine that the cell philapodia respond to a greater extent to the chemical gradient than the alignment of the fibers.94
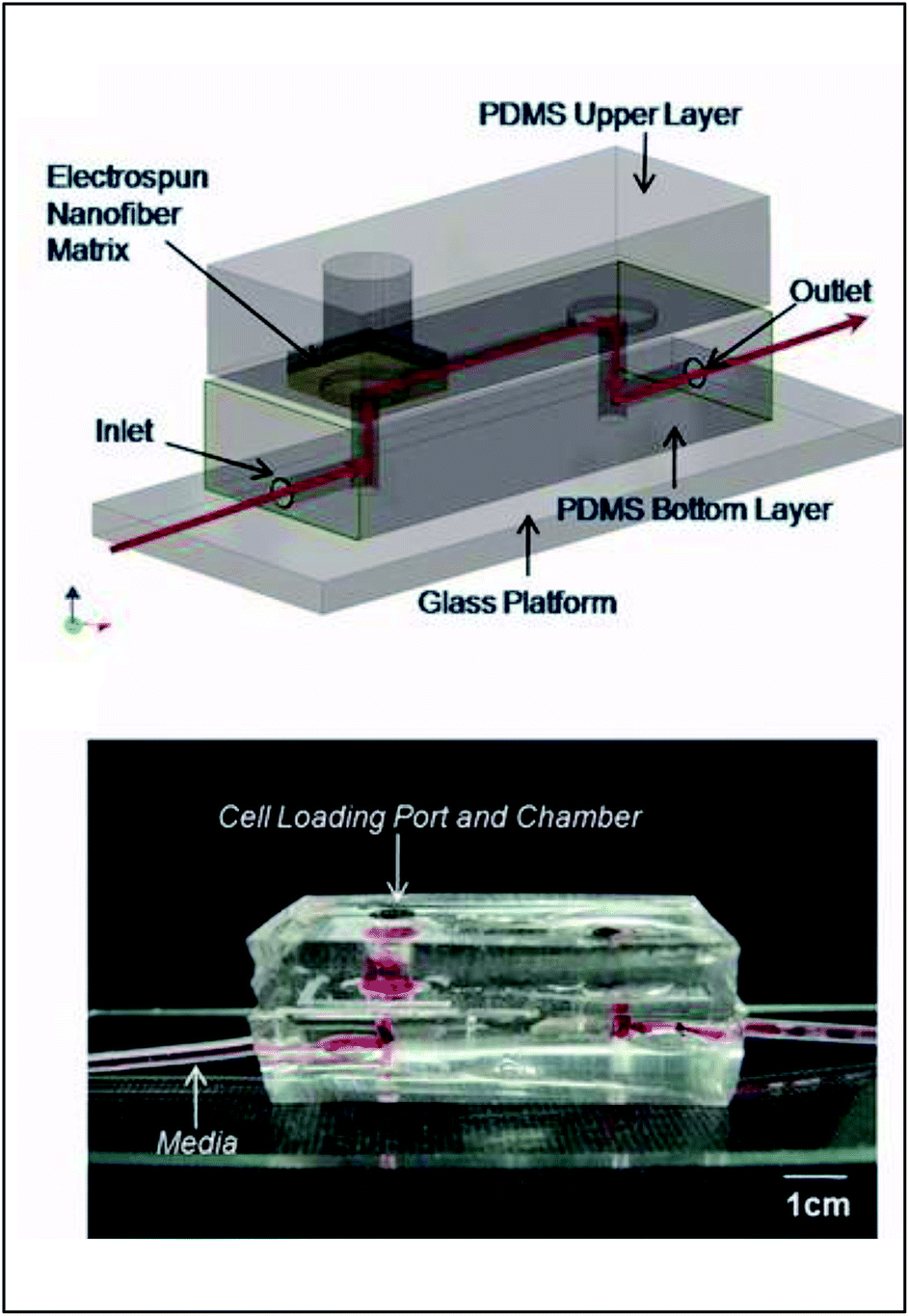 | ||
| Fig. 9 PDMS-based device for the incorporation of electrospun fiber scaffolds. (Top) Design representation of the chip. Red lines represent the path of fluid flow. (Bottom) Picture of the microchip with fibers and solution loaded into the cell loading port and the channel. (Adapted from ref. 93 with permission from John Wiley and Sons). | ||
 | ||
| Fig. 10 Method for incorporating multiple electrospun scaffolds into a microfluidic channel. (Top) (1) First fiber scaffold (polyurethane) is electrospun onto the substrate. (2) The substrate is covered with a mask and irradiated with deep ultraviolet light promoting crosslinking of the fibers. (3) The non-crosslinked fibers are dissolved away in THF followed by adeionized water rinse. (4) The substrate is dried. (5) Steps 1–4 are repeated for each additional fiber condition of interest. (6) A PDMS cover is aligned and sealed onto the glass substrate. (7) Completed device. (Bottom) False color fluorescence micrograph of 3T3 fibroblast cells culture in the microfluidic channel on the electrospun fibers. Blue-DNA are stained with DAP; red-actin filaments stained with Alexa Fluor 555 phalloidin; green is auto fluorescence of the fibers. (Adapted from ref. 94 with permission from AIP Publishing). | ||
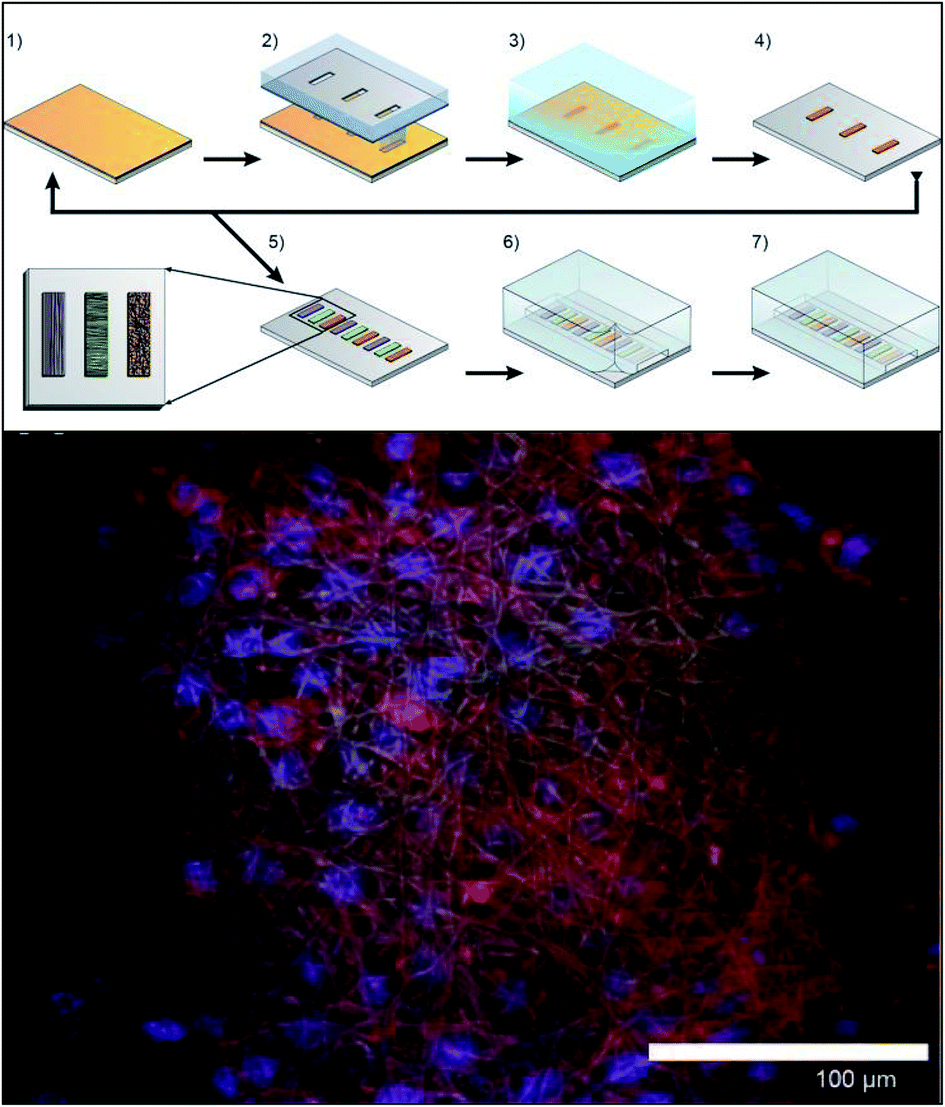
More recently Chen et al. explored ways to incorporate fiber scaffolds into a 3D printed microfluidic system,24,95,96 starting with an electrospinning approach to directly coat the inside of a channel with polycaprolactone fibers.95 Since this device was 3D printed in a hard plastic, printed threads could be included to host standard fittings making it easy to connect tubing to the device. The channels of the 3D printed device were coated with either fibers or protein to compare 2D vs. 3D culture of macrophages.95 After allowing macrophages growth for 24 hours, the cells were subjected to a circulating stimulation solution (LPS) for 12 hours followed by measurement of cytokine secretion (VEGF and IL-6). They determined that macrophages cultured on a 3D matrix secreted significantly more cytokines than those on a 2D matrix on a per cell basis. This same group has also reported a blow spinning set up that produced fiber scaffolds on an insert that could be fitted into a 3D printed flow channel.96 Chen et al. reported a second technique utilizing electrospinning that used a removable insert covered in fibers (Fig. 11).24 This device used two inserts stacked on top of each other that were covered with fibers (polystyrene, polycaprolactone, or silk) to make a fluidic channel (Fig. 11B). This device enabled cell culture outside of the device statically and then placed in the device for stimulation, then removed again for additional analysis.24 This cell culture module was coupled to a PDMS-based analytical module equipped with an optical fiber for analyzing nitrite via the Griess reaction (discussed in the next section). Additionally, the researchers were able to image large clusters a macrophage cells, another indicator of in vivo like cell growth (Fig. 11C).
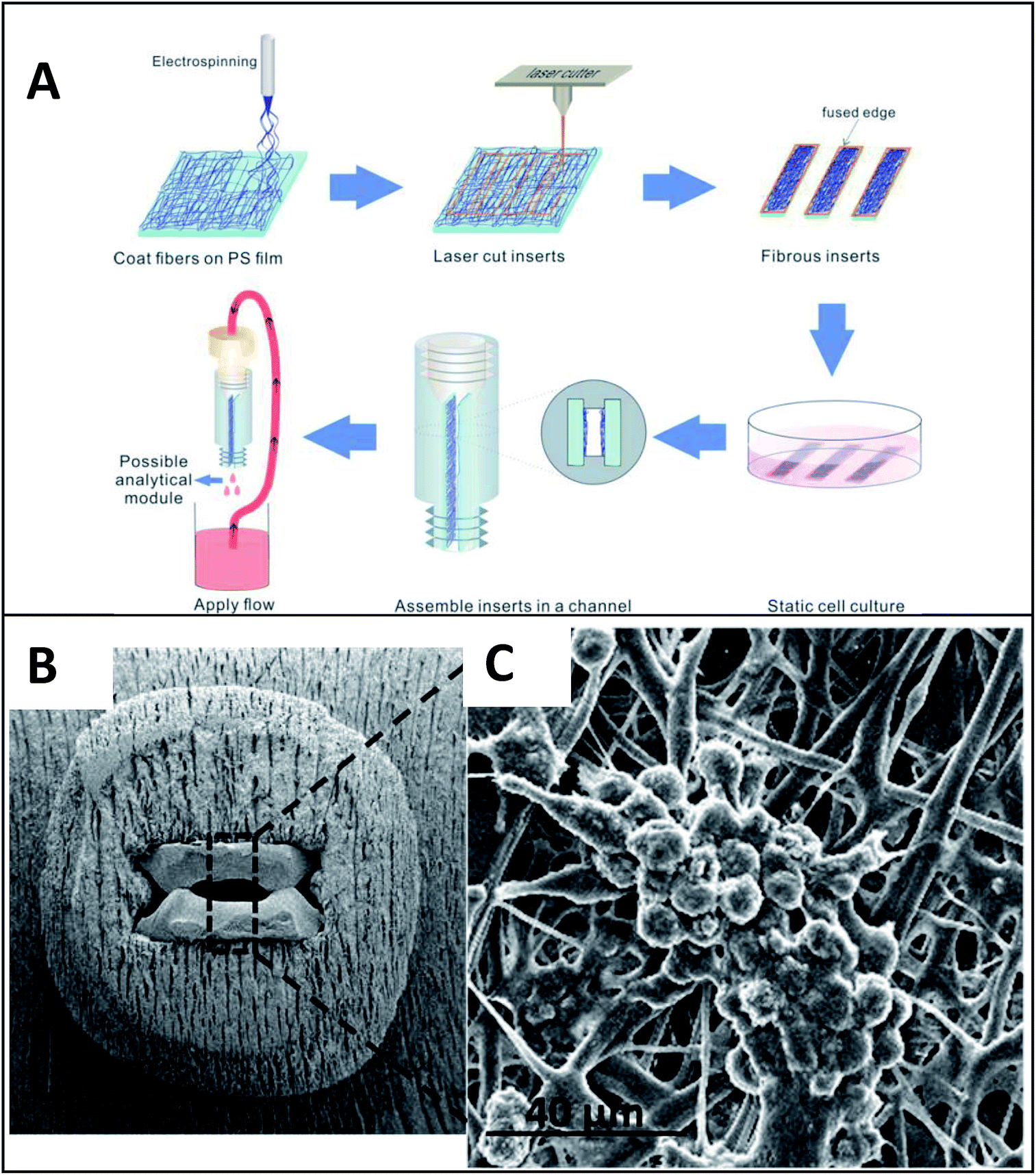 | ||
| Fig. 11 Insert-based fiber scaffold for microfluidic system. (A) Fibers are electrospun on a polystyrene film, followed by laser cutting of the insert to the desired dimensions. Cells can then be statically cultured on inserts, which can be placed into a 3D printed microfluidic device. The threaded connections allow for easy attachment to tubing, other cell culture modules, or analysis modules. (B). SEM of the polystyrene inserts places in a 3D printed cell culture module. (C) SEM of macrophage cells on a fibrous network (forming giant cell clusters). (Adapted from ref. 24 with permission from Springer Nature). | ||
Analysis in microfluidic-based 3D cell culture systems
An often-ignored factor in microfluidic-based cell culture is the manner in which key chemical messengers or signaling molecules can be detected to gain insights into communication pathways. These types of measurements are especially important in actively changing systems. The desire to integrate detection schemes also informs the fluidic design, as in some cases channel volume and possible dilution needs to be controlled to achieve reasonable detection limits. The following section outlines some of the approaches used for detection of key molecules in microfluidic-based 3D culture.Imaging
The vast majority of analysis coupled with microfluidic cell culture has involved imaging-based techniques. This includes simple cell counting methods to determine proliferation rates through complex staining protocols coupled with confocal microscopy to evaluate cell viability and cell morphology.50,58,77,94,97,98 The overwhelming conclusion is that the presence of these complex extracellular matrices affect growth rates, cell morphology, and provides more in vivo like cell performance.One interesting application of imaging-based analysis comes from Bauer et al., where a 3D collagen gel was placed in a microchannel, with breast carcinoma cells being grown under either monoculture or co-culture (with human mammary fibroblasts) conditions. The total cell area was measured and it was found that after 5 days of co-culture, the carcinoma cell clusters were larger in size than those seen in mono-culture conditions (Fig. 12).58 The same group was able to stain for F-actin and Tubulin, proteins important to cell mobility, to monitor neutrophil polarization on 2D and 3D substrates within a microfluidic device.99 They were able to show that on a 2D surface the cell displayed a polarized morphology with a single broad leading edge (Fig. 13) while a 3D hydrogel environment forces the cell to have multiple leading edge pseudopods that aid in mobility.99
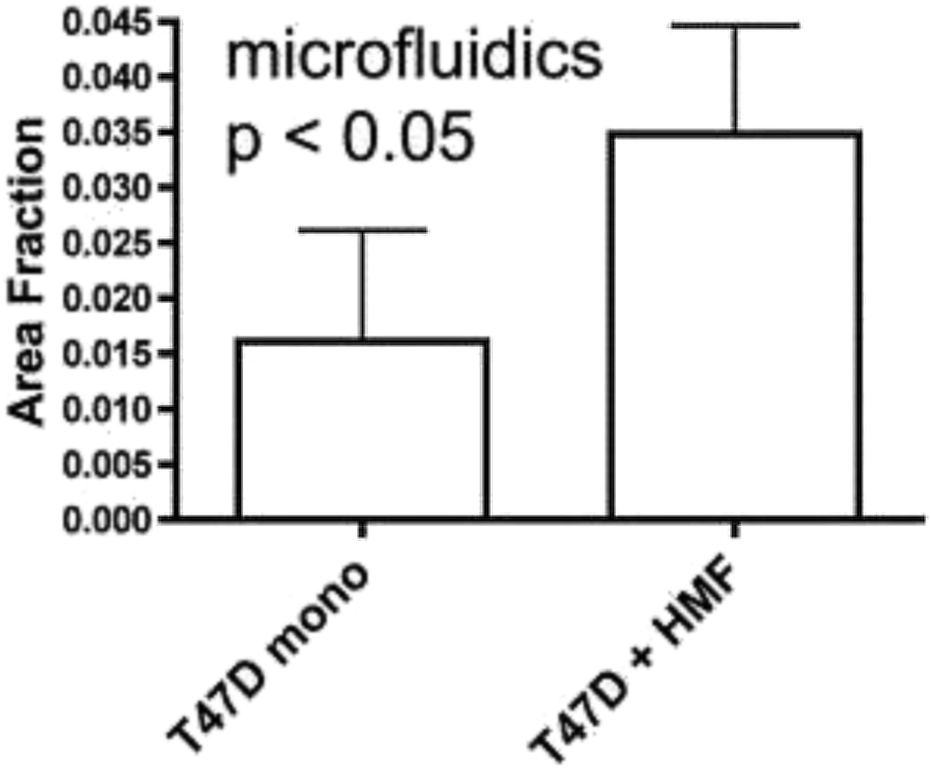 | ||
| Fig. 12 Using fluorescence microscopy, the total area of tumor cell clusters were measured and compared under mono-culture and co-culture conditions. It was found that T47D cells co-cultured with HMF are bigger in size than monocultured cells. (Adapted from ref. 58 with permission from Oxford University Press). | ||
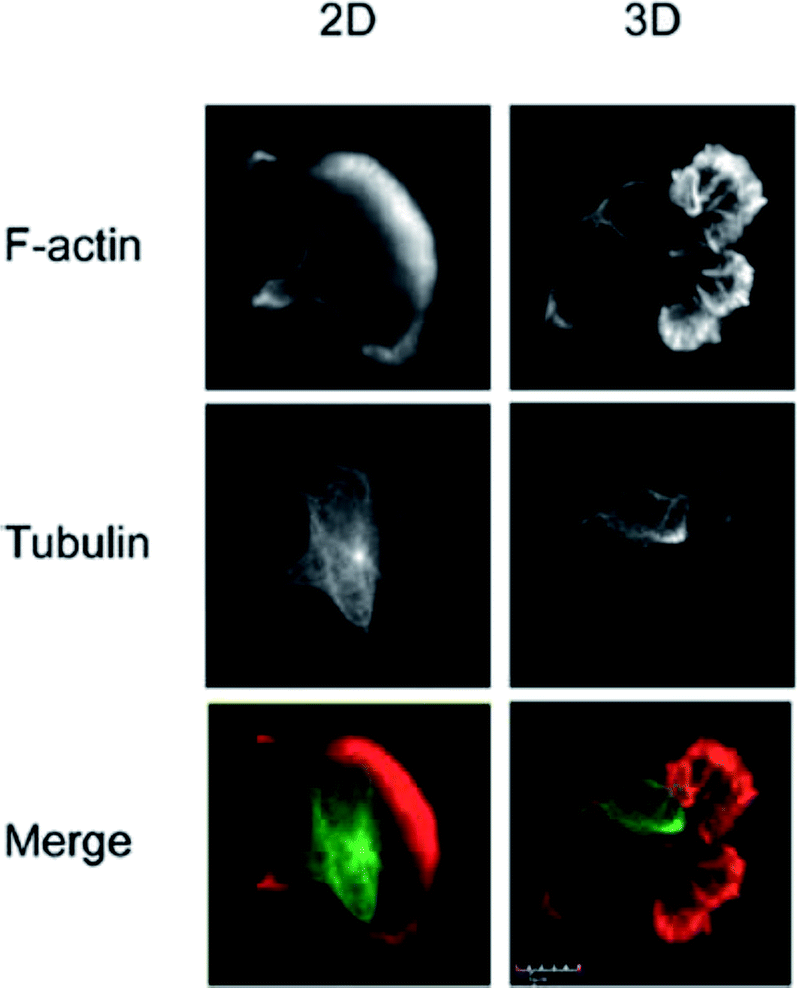 | ||
| Fig. 13 Neutrophil polarization in 2D and 3D culture. Neutrophils were stained for F-actin (red) and tubulin (green) to determine the morphology of cells on a 2D fibrinogen coated channel vs. a channel with a hydrogel. It was determined that the 2D substrate lead to a polarized morphology with a single broad leading edge, while the 3D hydrogel environment yielded multiple pseudo-pods. (Adapted from ref. 99 with permission from Springer Nature). | ||
Using a stacked paper based 3D culture platform Mosadegh et al. were able to use fluorescent imaging to map lung cancer cell migration as it is effected by oxygen gradients.77 GFP-expressing cells were imaged on each layer of the stack after 24 hours for the following gas and media accessible conditions (Fig. 14): gas/media access on top of the stack with cells on the top layer; gas/media access on top of the stack with cells on the bottom layer; gas/media top and bottom with cells in the middle layer; gas access on the top and bottom layers with media being accessible only to the top layer and cells in the center layer. Utilizing a PDMS layer at the bottom of the stack (to allow only gas exchange, Fig. 14D) versus an opening at the bottom of the stack (to allow both gas and media exchange, Fig. 14C) and seeding the cells in the center of the stack, it was determined that the cells migrated bidirectionally towards the ends of the stack. Since the migration profile was the same in both cases, it was concluded that gas exchange, not media exchange, is the primary factor in cell migration.77
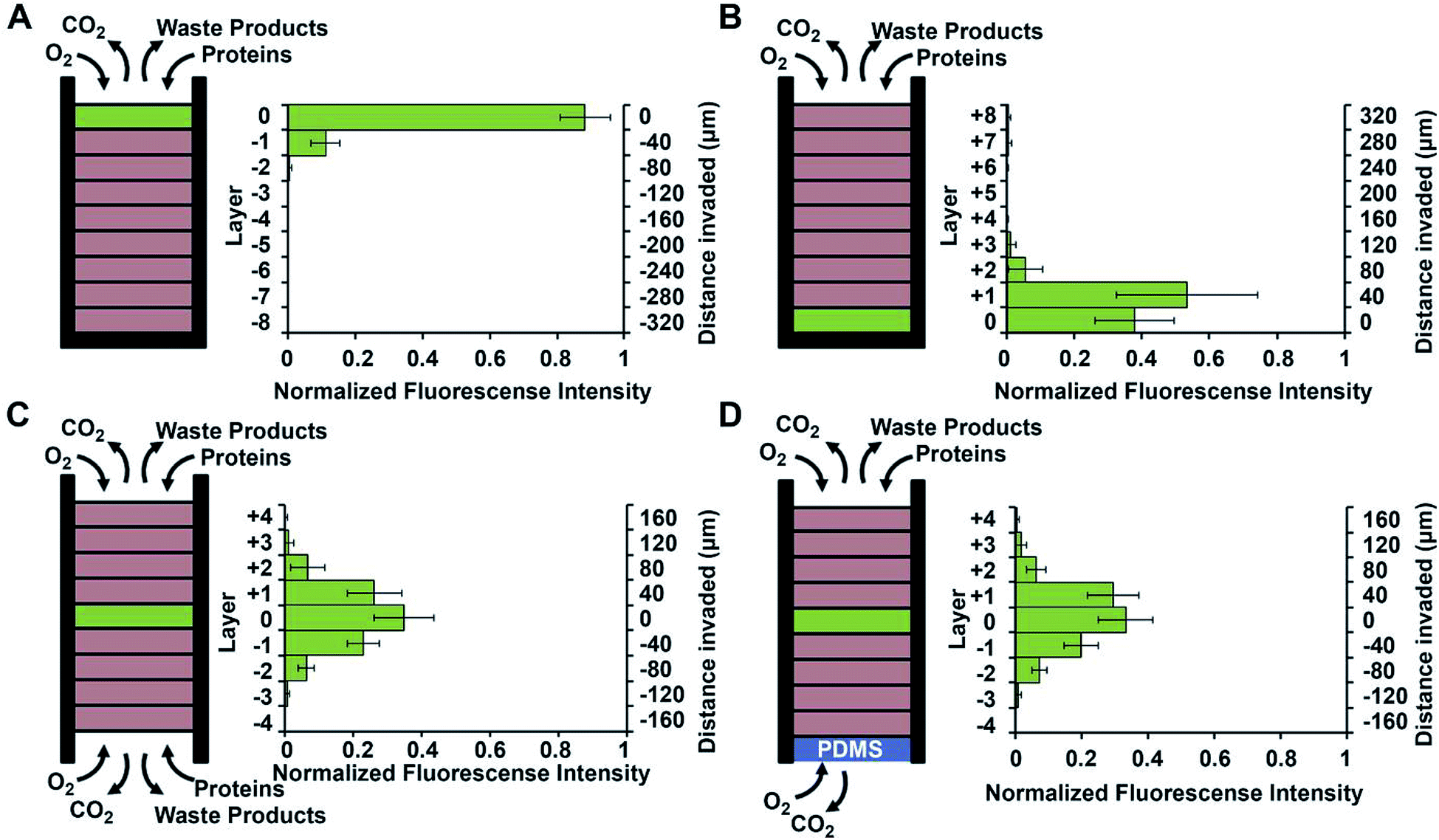 | ||
| Fig. 14 Evidence of chemotaxis in a sub-population of A549-HGF-M cells (lung cancer cells). Cells were cultured and stacked similar to the system seen in Fig. 8 except a hard plastic shell was placed around the stack to further limit the diffusion of nutrients and waste products. (A) Cells were positioned at the top of the stack close to the source of oxygen and little movement was seen. (B) Cells were positioned at the bottom of the stack furthest away from the oxygen and a large amount of chemotaxis was seen. (C) Cell were positioned in the center of the stack and media was allowed to penetrate from both side of the stack. Symmetric movement was seen. (D) The cells were placed in the center of the stack once again but a PDMS gas permeable membrane was used to close off the bottom. Symmetric movement was again seen confirming that chemotaxis is a result of available oxygen. (Adapted from ref. 77 with permission from Elsevier). | ||
Offline detection
Some work has been done coupling offline detection schemes to microfluidic 3D cell culture. One issue with off-line analysis is the need to collect enough sample so that it can be physically pipetted or transferred from the device. One analysis technique that is compatible with small sample volumes is enzyme-linked immunosorbent assays (ELISA). Kim et al. took this approach to quantify the secretion of albumin and alpha-fetoprotein from HepG2 cells, collecting 2.4 mL samples every 2 days for 14 days total.100 This time between sample collection limited the temporal resolution but maximized the number of replicates that could be made and the number of proteins that could be analyzed (up to 12). Other common methods include the lysis of the cells and subsequent RNA analysis or quantitative PCR analysis.95,100–102Another approach for offline analysis comes is the use of MALDI imaging mass spectrometry as reported by LaBonia et al.103 In this instance, tumor spheroids were cultured in a transwell insert above a fluidic channel. This allowed dosage of the spheroid that could be removed later for analysis. Using MALDI-IMS the entire tumor cluster could be mapped for both the drug and the active metabolite, allowing a more complete picture of the effects of dosing on the spheroid system.
Online detection
The advantages of on-line detection include the ability to continuously monitor the system of interest (improving temporal resolution) and minimizing dilution effects that are often seen in off-line detection schemes. The ability to integrate online detection depends greatly on the scaffold material chosen. Paper-based analysis of small molecules and proteins is becoming common place but has not been widely coupled to cell culture.73 One such example comes from Lei et al., who used a paper substrate for the cell co-culture then fluidically connected the cell growth areas with a hydrogel channel layer.101 As part of their analysis it was determined cell phosphorylation solutions using a commonly used colorimetric immunoassay could be pipetted directly on the cell culture areas. While this is only one example of direct colorimetric analysis coupled to 3D cell culture, the area of paper based colorimetric analysis is robust and well-reviewed.104More recently advances have been made on in-line detection of analytes relevant to 3D cell culture in a more modular approach. In 2018 Chen et al. developed an insert-based 3D printed device in which a fiber scaffold with cultured cells could be easily inserted and removed.24 This device had a threaded component on either end such that tubing could be easily attached. This allowed connection of the cell module to a flow-based absorbance system (Fig. 15A). Using the cell module attached to a mixing T, Griess reagent was mixed in-line with the cell effluent. This mixed solution then flowed through a PDMS-device that was fitted with two optical fibers on either side of the channel, one fiber being connected to a tungsten lamp and the other leading to a photodiode array. The PDMS-based absorbance module was able to detect the colorimetric product from the Griess reaction with nitrite (a marker of the M1 macrophage phenotype). Using this set up they were able to determine that stimulated macrophages cultured on a 3D matrix rather than a 2D matrix can be promoted to the M1 state in a more in vivo-like timescale.
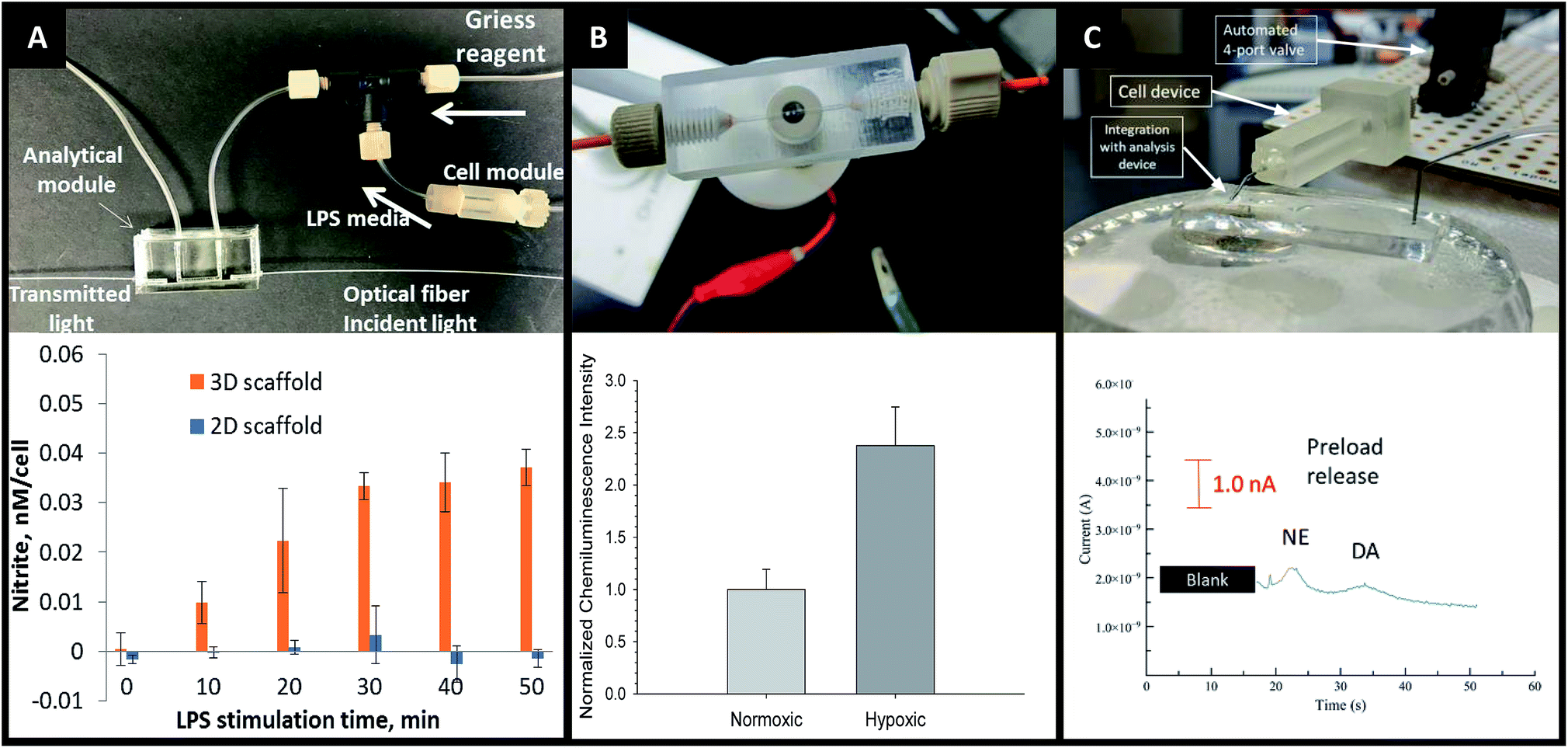 | ||
| Fig. 15 3D printed devices as a mechanism for cell culture and analysis. (A) A 3D printed cell culture module with fiber scaffold inserts is connected to a PDMS-based optical flow cell. Using this set up nitrate from macrophages was quantified. It was determined that macrophages on 3D scaffolds better mimic the in vivo M1 state. (Adapted from ref. 24 with permission from Springer Nature). (B) A 3D printed electrochemical flow cell with threaded inlet and outlet for easy capillary connections. Using these threaded electrodes the researchers were able to confirm normoxic or hypoxic conditions in flowing red blood cells while simultaneously measuring ATP release with the luciferin/luciferase assay. (Adapted from ref. 39 with permission from The Royal Society of Chemistry). (C) A 3D printed cell culture module is interfaced with PDMS chip by flow matching the solution being pushed through the cell culture module and the fluid being pulled through the PDMS chip. Using this technique, a 3D printed cell culture module could be coupled with a PDMS-based electrophoresis chip with electrochemical detection to separate and quantify norepinephrine and dopamine release from PC12 cells. (Adapted from ref. 41 with permission from The Royal Society of Chemistry). | ||
By utilizing electrodes fabricated in threaded fittings, Erkal et al. were able to electrochemically quantify oxygen and nitric oxide levels in a 3D printed fluidic device (Fig. 15B).39 The researchers were able to develop a fabrication technique in which multiple electrode materials (Au, Ag/AgCl, Pd, Pt) could be embedded into a commercially available PEEK fitting, which could be threaded into a fluidic device as desired. These devices were used to show the feasibility of measuring dopamine and nitric oxide standards, as well as oxygen in a flowing stream of red blood cells.39 Using similar electrode materials but with a wall-jet electrode and the previously mentioned insert-based 3D fiber/cell culture platform, Munshi et al. were able to quantify nitric oxide release from ATP-stimulated endothelial cells.105
Mehl et al. later reported a similar fiber-based cell culture device that could be interfaced to traditional PDMS-based analysis chips (Fig. 15C).41 Ultimately, they were able to interface a cell culture module with a microchip-based electrophoresis PDMS/valving chip with integrated electrochemical detection for detection of both norepinephrine and dopamine release from PC12 cells.41 This approach used 3 syringe pumps, 2 for the attrition of either buffer or a stimulating solution on the cell culture side and 1 to withdraw solution through a PDMS electrophoresis pump. The interface between the two devices was a small reservoir droplet. By matching the flow rates of the pump, cell release solutions could be coupled to an electrophoretic separation technique (with injections via on-chip valves). It was found that two analytes released from PC 12 cells (dopamine and norepinephrine) could be simultaneously separated and detected in near real-time (Fig. 15C).41
Conclusions
We have presented an overview of the work being performed in the combined areas of 3D cell culture, microfluidics, and integrated analysis. There are four main approaches when it comes to 3D cell culture (suspension, hydrogel, paper, and fiber), all with applications that can be integrated with microfluidic techniques; however, joining of the two techniques has been relatively slow in the community. Clear advantages have been demonstrated when attempting to mimic in vivo environments and complex cell-to-cell interactions. It is clear that microfluidic-based 3D cell culture coupled with analysis can greatly improve our ability to mimic and understand in vivo systems.Future directions can focus on better characterization of these complex cell-to-ECM and cell-to-cell interactions. While the majority of the analysis has focus on cell morphology such as alignment, migration, or proliferation rates, this yields an incomplete picture. It has been repeatedly shown that all of these parameters affect cell signaling pathways that are not being fully explored. Another area for future exploration is the quantification of small molecule uptake and release as well as protein expression. The limited work done thus far often lacks temporal resolution so that measurements are on time scales relevant to cell function. Real-time analysis is a must for understanding these complex reactions and the microfluidic design needs to be informed by the method of detection integration. We expect the intermingling of complex ECMs with microfluidic cell culture and analysis to be a robust research area moving forward.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support from the National Institutes of Health (NS105888) is acknowledged.References
- R. G. Harrison, M. J. Greenman, F. P. Mall and C. M. Jackson, Anat. Rec., 1907, 1, 116–128 CrossRef.
- R. C. Parker, Methods of Tissue Culture, 1938 Search PubMed.
- F. Jacoby and S. J. Darke, Nature, 1948, 161, 768–769 CrossRef CAS PubMed.
- L. L. Coriell, A. E. Greene and R. K. Silver, Cryobiology, 1964, 1, 72–79 CrossRef.
- D. Barnes and G. Sato, Cell, 1980, 22, 649–655 CrossRef CAS PubMed.
- K. Gurvinder and J. Dufour, Spermatogenesis, 2012, 2, 1–5 CrossRef PubMed.
- M. Kapalczynska, T. Kolenda, W. Przybyla, M. Zajaczkowska, A. Teresiak, V. Filas, M. Ibbs, R. Blizniak, L. Luczewski and K. Lamperska, Arch. Med. Sci., 2018, 14, 910–919 Search PubMed.
- R. R. Mitry and R. D. Hughes, Human Cell Culture Protocols, 2012 Search PubMed.
- C. F. Amstein and P. A. Hartman, J. Clin. Microbiol., 1975, 2, 46–54 CAS.
- W. S. Ramsey, W. Hertl, E. D. Nowlan and N. J. Binkowski, In Vitro, 1984, 20, 802–808 CrossRef CAS PubMed.
- M. Shen and T. A. Horbett, J. Biomed. Mater. Res., 2001, 57, 336–345 CrossRef CAS PubMed.
- K. Duval, H. Grover, L. H. Han, Y. Mou, A. F. Pegoraro, J. Fredberg and Z. Chen, Physiology, 2017, 32, 266–277 CrossRef CAS PubMed.
- D. Kosoff, J. Yu, V. Suresh, D. J. Beebe and J. M. Lang, Lab Chip, 2018, 18, 3011–3017 RSC.
- J. W. Haycock, 3D Cell Culture, Humana Press, 2011 Search PubMed.
- D. Huh, G. A. Hamilton and D. E. Ingber, Trends Cell Biol., 2011, 21, 745–754 CrossRef CAS PubMed.
- A. Dhaliwal, Mater. Methods, 2012, 2, 162 Search PubMed.
- M. Mirbagheri, V. Adibnia, B. R. Hughes, S. D. Waldman, X. Banquy and D. K. Hwang, Mater. Horiz., 2019, 6, 45–71 RSC.
- D. J. Maltman and S. A. Przyborski, Biochem. Soc. Trans., 2010, 38, 1072–1075 CrossRef CAS PubMed.
- M. W. Tibbitt and K. S. Anseth, Biotechnol. Bioeng., 2009, 103, 655–663 CrossRef CAS PubMed.
- J. N. Hanson Shepherd, S. T. Parker, R. F. Shepherd, M. U. Gillette, J. A. Lewis and R. G. Nuzzo, Adv. Funct. Mater., 2011, 21, 47–54 CrossRef CAS PubMed.
- N. Pupinyo, M. Chatatikun, A. Chiabchalard and W. Laiwattanapaisal, Analyst, 2018, 144, 290–298 RSC.
- S. Chung, G. J. Lim and J. Y. Lee, Sci. Rep., 2019, 9, 1–9 CrossRef CAS PubMed.
- S. Breslin and L. O'Driscoll, Drug Discovery Today, 2013, 18, 240–249 CrossRef CAS PubMed.
- C. Chen, A. D. Townsend, E. A. Hayter, H. M. Birk, S. A. Sell and R. S. Martin, Anal. Bioanal. Chem., 2018, 410, 3025–3035 CrossRef CAS PubMed.
- S. K. Sia and G. M. Whitesides, Electrophoresis, 2003, 24, 3563–3576 CrossRef CAS PubMed.
- S. Torino, B. Corrado, M. Iodice and G. Coppola, Inventions, 2018, 3, 1–14 CrossRef.
- Y. S. Heo, L. M. Cabrera, J. W. Song, N. Futai, Y. C. Tung, G. D. Smith and S. Takayama, Anal. Chem., 2007, 79, 1126–1134 CrossRef CAS PubMed.
- K. J. Regehr, M. Domenech, J. T. Koepsel, K. C. Carver, S. J. Ellison-Zelski, W. L. Murphy, L. A. Schuler, E. T. Alarid and D. J. Beebe, Lab Chip, 2009, 9, 2132–2139 RSC.
- B. Gale, A. Jafek, C. Lambert, B. Goenner, H. Moghimifam, U. Nze and S. Kamarapu, Inventions, 2018, 3, 1–25 CrossRef.
- S. Waheed, J. M. Cabot, N. P. Macdonald, T. Lewis, R. M. Guijt, B. Paull and M. C. Breadmore, Lab Chip, 2016, 16, 1993–2013 RSC.
- C. Chen, B. T. Mehl, A. S. Munshi, A. D. Townsend, D. M. Spence and R. S. Martin, Anal. Methods, 2016, 8, 6005–6012 RSC.
- B. C. Gross, J. L. Erkal, S. Y. Lockwood, C. Chen and D. M. Spence, Anal. Chem., 2014, 86, 3240–3253 CrossRef CAS PubMed.
- A. D. Castiaux, C. W. Pinger, E. A. Hayter, M. E. Bunn, R. S. Martin and D. M. Spence, Anal. Chem., 2019, 91, 6910–6917 CAS.
- Y. Gao, D. Majumdar, B. Jovanovic, C. Shaifer, P. C. Lin, A. Zijlstra, D. J. Webb and D. Li, Biomed. Microdevices, 2011, 13, 539–548 CrossRef CAS PubMed.
- S. Knowlton, C. H. Yu, F. Ersoy, S. Emadi, A. Khademhosseini and S. Tasoglu, Biofabrication, 2016, 8, 1–13 CrossRef PubMed.
- S. Halldorsson, E. Lucumi, R. Gomez-Sjoberg and R. M. T. Fleming, Biosens. Bioelectron., 2015, 63, 218–231 CrossRef CAS PubMed.
- J. Sibbitts, K. A. Sellens, S. Jia, S. A. Klasner and C. T. Culbertson, Anal. Chem., 2018, 90, 65–85 CrossRef CAS PubMed.
- X. Wang, L. Yi, N. Mukhitov, A. M. Schrell, R. Dhumpa and M. G. Roper, J. Chromatogr. A, 2015, 1382, 98–116 CrossRef CAS PubMed.
- J. L. Erkal, A. Selimovic, B. C. Gross, S. Y. Lockwood, E. L. Walton, S. McNamara, R. S. Martin and D. M. Spence, Lab Chip, 2014, 14, 2023–2032 RSC.
- C. E. Dugan, J. P. Grinias, S. D. Parlee, M. El-Azzouny, C. R. Evans and R. T. Kennedy, Anal. Bioanal. Chem., 2017, 409, 169–178 CrossRef CAS PubMed.
- B. T. Mehl and R. S. Martin, Anal. Methods, 2019, 11, 1064–1072 RSC.
- N. A. Lacher, S. M. Lunte and R. S. Martin, Anal. Chem., 2004, 76, 2482–2491 CrossRef CAS PubMed.
- S. J. Collins, R. C. Gallo and R. E. Gallagher, Nature, 1977, 270, 347–349 CrossRef CAS PubMed.
- J. Wu, G. King, A. J. Daugulis, P. Faulkner, D. H. Bone and M. F. A. Goosen, Appl. Microbiol. Biotechnol., 1989, 32, 249–255 CrossRef CAS.
- L. Chu and D. K. Robinson, Curr. Opin. Biotechnol., 2001, 12, 180–187 CrossRef CAS PubMed.
- A. Naivs, The Embryo Project Encyclopedia, 2007 Search PubMed.
- R. Foty, J. Visualized Exp., 2011, 51, 1–4 Search PubMed.
- M. Shri, H. Agrawal, P. Rani, D. Singh and S. K. Onteru, Sci. Rep., 2017, 7, 1–14 CrossRef CAS PubMed.
- H. W. Wu, Y. H. Hsiao, C. C. Chen, S. F. Yet and C. H. Hsu, Molecules, 2016, 21, 1–11 Search PubMed.
- O. Frey, P. M. Misun, D. A. Fluri, J. G. Hengstler and A. Hierlemann, Nat. Commun., 2014, 5, 1–11 Search PubMed.
- S. R. Caliari and J. A. Burdick, Nat. Methods, 2016, 13, 405–414 CrossRef CAS PubMed.
- N. Chirani, L. H. Yahia, L. Gritsch, F. L. Motta, S. CHirani and S. Fare, J. Biomed. Sci. Res., 2016, 4, 1–23 Search PubMed.
- M. C. Cushing and K. S. Anseth, Science, 2007, 316, 1133–1134 CrossRef CAS PubMed.
- E. E. Antoine, P. P. Vlachos and M. N. Rylander, Tissue Eng., Part B, 2014, 20, 683–696 CrossRef CAS PubMed.
- I. Meyvantsson, J. W. Warrick, S. Hayes, A. Skoien and D. J. Beebe, Lab Chip, 2008, 8, 717–724 RSC.
- G. Walker and D. J. Beebe, Lab Chip, 2002, 2, 131–134 RSC.
- E. W. Young and D. J. Beebe, Chem. Soc. Rev., 2010, 39, 1036–1048 RSC.
- M. Bauer, G. Su, D. J. Beebe and A. Friedl, Integr. Biol., 2010, 2, 371–378 CrossRef PubMed.
- B. Burkel, B. A. Morris, S. M. Ponik, K. M. Riching, K. W. Eliceiri and P. J. Keely, J. Visualized Exp., 2016, 111, 1–7 Search PubMed.
- S. Chung, R. Sudo, P. J. Mack, C. R. Wan, V. Vickerman and R. D. Kamm, Lab Chip, 2009, 9, 269–275 RSC.
- L. L. Bischel, B. P. Casavant, P. A. Young, K. W. Eliceiri, H. S. Basu and D. J. Beebe, Integr. Biol., 2014, 6, 627–635 CrossRef CAS PubMed.
- H. Li, C. Tan and L. Li, Mater. Des., 2018, 159, 20–38 CrossRef CAS.
- Y. He, F. Yang, H. Zhao, Q. Gao, B. Xia and J. Fu, Sci. Rep., 2016, 6, 1–13 CrossRef PubMed.
- S. You, J. Li, W. Zhu, C. Yu, D. Mei and S. Chen, J. Mater. Chem. B, 2018, 6, 2187–2197 RSC.
- N. Contessi Negrini, L. Bonetti, L. Contili and S. Farè, Bioprinting, 2018, 10, 1–10 CrossRef.
- R. Derda, A. Laromaine, A. Mammoto, S. K. Tang, T. Mammoto, D. E. Ingber and G. M. Whitesides, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18457–18462 CrossRef CAS PubMed.
- K. Ng, B. Gao, K. W. Yong, Y. Li, M. Shi, X. Zhao, Z. Li, X. Zhang, B. Pingguan-Murphy, H. Yang and F. Xu, Mater. Today, 2017, 20, 32–44 CrossRef CAS.
- X. Wu, S. Suvarnapathaki, K. Walsh and G. Camci-Unal, MRS Commun., 2018, 8, 1–14 CrossRef.
- S. K. Mahadeva, K. Walus and B. Stoeber, ACS Appl. Mater. Interfaces, 2015, 7, 8345–8362 CrossRef CAS PubMed.
- E. Carrilho, A. W. Martinez and G. M. Whitesides, Anal. Chem., 2009, 81, 7091–7095 CrossRef CAS PubMed.
- A. W. Martinez, S. T. Phillips and G. M. Whitesides, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 19606–19611 CrossRef CAS PubMed.
- T. Akyazi, L. Basabe-Desmonts and F. Benito-Lopez, Anal. Chim. Acta, 2018, 1001, 1–17 CrossRef CAS PubMed.
- A. W. Martinez, S. T. Phillips, M. J. Butte and G. M. Whitesides, Angew. Chem., 2007, 46, 1318–1320 CrossRef CAS PubMed.
- F. F. Tao, X. Xiao, K. F. Lei and I. C. Lee, BioChip J., 2015, 9, 97–104 CrossRef CAS.
- J. A. Matthews, G. E. Wnek, D. G. Simpson and G. L. Bowlin, Biomacromolecules, 2002, 3, 232–238 CrossRef CAS PubMed.
- Y. Lu, W. Shi, L. Jiang, J. Qin and B. Lin, Electrophoresis, 2009, 30, 1497–1500 CrossRef CAS PubMed.
- B. Mosadegh, M. R. Lockett, K. T. Minn, K. A. Simon, K. Gilbert, S. Hillier, D. Newsome, H. Li, A. B. Hall, D. M. Boucher, B. K. Eustace and G. M. Whitesides, Biomaterials, 2015, 52, 262–271 CrossRef CAS PubMed.
- R. M. Kenney, M. W. Boyce, A. S. Truong, C. R. Bagnell and M. R. Lockett, Analyst, 2016, 141, 661–668 RSC.
- R. M. Kenney, M. W. Boyce, N. A. Whitman, B. P. Kromhout and M. R. Lockett, Anal. Chem., 2018, 90, 2376–2383 CrossRef CAS PubMed.
- M. Alazaba, G. R. Mitchellb, F. J. Davisd and S. D. Mohand, Procedia Manufacturing, 2017, 12, 66–78 CrossRef.
- J. D. Schiffman and C. L. Schauer, Polym. Rev., 2008, 48, 317–352 CrossRef CAS.
- N. E. Zander, J. Appl. Polym. Sci., 2015, 132, 41269 CrossRef.
- S. Chung, N. P. Ingle, G. A. Montero, S. H. Kim and M. W. King, Acta Biomater., 2010, 6, 1958–1967 CrossRef CAS PubMed.
- E. S. Medeiros, G. M. Glenn, A. P. Klamczynski, W. J. Orts and L. H. C. Mattoso, J. Appl. Polym. Sci., 2009, 113, 2322–2330 CrossRef CAS.
- J. L. Daristotle, A. M. Behrens, A. D. Sandler and P. Kofinas, ACS Appl. Mater. Interfaces, 2016, 8, 34951–34963 CrossRef CAS.
- M. K. Leach, Z. Q. Feng, S. J. Tuck and J. M. Corey, J. Visualized Exp., 2011, 47, 1–4 Search PubMed.
- T. Liu, W. K. Teng, B. P. Chan and S. Y. Chew, J. Biomed. Mater. Res., Part A, 2010, 95, 276–282 CrossRef PubMed.
- J. Xue, J. Xie, W. Liu and Y. Xia, Acc. Chem. Res., 2017, 50, 1976–1987 CrossRef CAS PubMed.
- B. S. Jha, C. E. Ayres, J. R. Bowman, T. A. Telemeco, S. A. Sell, G. L. Bowlin and D. G. Simpson, J. Nanomater., 2011, 2011, 1–15 CrossRef.
- Y. H. Jin Nam, S. Agarwal and J. Lannutti, J. Appl. Polym. Sci., 2007, 107, 1547–1554 Search PubMed.
- A. Elamparithi, A. M. Punnoose and S. Kuruvilla, Artif. Cells, Nanomed., Biotechnol., 2016, 44, 1318–1325 CAS.
- B. Dong, O. Arnoult, M. E. Smith and G. E. Wnek, Macromol. Rapid Commun., 2009, 30, 539–542 CrossRef CAS PubMed.
- K. H. Lee, G. H. Kwon, S. J. Shin, J. Y. Baek, D. K. Han, Y. Park and S. H. Lee, J. Biomed. Mater. Res., Part A, 2009, 90, 619–628 CrossRef PubMed.
- P. Wallin, C. Zanden, B. Carlberg, N. Hellstrom Erkenstam, J. Liu and J. Gold, Biomicrofluidics, 2012, 6, 1–18 CrossRef.
- C. Chen, B. T. Mehl, S. A. Sell and R. S. Martin, Analyst, 2016, 141, 5311–5320 RSC.
- C. Chen, A. D. Townsend, S. A. Sell and R. S. Martin, Anal. Methods, 2017, 9, 3274–3283 RSC.
- A. P. Aijian and R. L. Garrell, J. Lab. Autom., 2015, 20, 283–295 CrossRef CAS PubMed.
- W. Yan, Q. Zhang, B. Chen, G.-T. Liang, W.-X. Li, X.-M. Zhou and D.-Y. Liu, Chin. J. Anal. Chem., 2013, 41, 822–827 CrossRef CAS.
- Y. Yamahashi, P. J. Cavnar, L. E. Hind, E. Berthier, D. A. Bennin, D. Beebe and A. Huttenlocher, Biomed. Microdevices, 2015, 17, 1–9 CrossRef CAS PubMed.
- J. H. Kim, J. Y. Park, S. Jin, S. Yoon, J. Y. Kwak and Y. H. Jeong, Nanomaterials, 2019, 9, 1–12 Search PubMed.
- K. F. Lei, C. H. Chang and M. J. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 13092–13101 CrossRef CAS PubMed.
- A. S. Truong, C. A. Lochbaum, M. W. Boyce and M. R. Lockett, Anal. Chem., 2015, 87, 11263–11270 CrossRef CAS PubMed.
- G. J. LaBonia, S. Y. Lockwood, A. A. Heller, D. M. Spence and A. B. Hummon, Proteomics, 2016, 16, 1814–1821 CrossRef CAS PubMed.
- E. W. Nery and L. T. Kubota, Anal. Bioanal. Chem., 2013, 405, 7573–7595 CrossRef CAS PubMed.
- A. S. Munshi, C. Chen, A. D. Townsend and R. S. Martin, Anal. Methods, 2018, 10, 3364–3374 RSC.
| This journal is © The Royal Society of Chemistry 2019 |