
Graphene oxide-circular aptamer based colorimetric protein detection on bioactive paper†
 *a
*a
Abstract
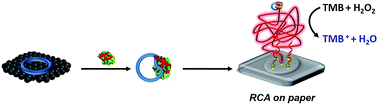
Paper-based sensor technology represents a new class of point-of-care (POC) diagnostic devices that is affordable, portable, rapid and scalable for manufacturing. One of the goals of developing new paper-based POC diagnostics is the need to recognize targets with high specificity and sensitivity. Herein, a graphene oxide-circular aptamer based assay is described for highly specific and sensitive detection of a target protein on bioactive paper. The circular aptamer was self-assembled onto graphene oxide to generate the biorecognition moieties, followed by the desorption reaction induced by the specific binding of the target. The released circular aptamer then hybridizes to paper-bound DNA primers, thus initiating the rolling circle amplification reaction to produce a long DNA molecule containing multiple horseradish peroxidase-mimicking DNAzyme units, which further catalyze the oxidation of substrates by H2O2 in the presence of hemin to yield a distinct colorimetric signal on paper. Under the optimal conditions, this dual amplification method can achieve detection of platelet-derived growth factor (PDGF) at a concentration as low as 100 pM. Overall, this strategy of paper-based nanobiosensors may hold great potential for rapid, accurate and inexpensive biomarker detection in POC applications.
- This article is part of the themed collection: Analytical Methods Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

