
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Microperoxidase-11 modified mesoporous SnO2 film electrodes for the detection of antimalarial drug artemisinin†
Lambros Aris
Ioannidis
,
Pavlos
Nikolaou
 ,
Apostolos
Panagiotopoulos
,
Andriana
Vassi
and
Emmanuel
Topoglidis
*
,
Apostolos
Panagiotopoulos
,
Andriana
Vassi
and
Emmanuel
Topoglidis
*
Department of Materials Science, University of Patras, 26504 Patras, Greece. E-mail: etop@upatras.gr
First published on 27th May 2019
Abstract
This work presents a simple and efficient preparation method for microperoxidase-11 (MP11) modified mesoporous SnO2 films on ITO glass substrates (MP11/DDAB/SnO2/ITO) for the development of a sensitive electrochemical drug sensor for the determination of antimalarial endoperoxide artemisinin (ART). Scanning Electron Microscopy (SEM), X-ray Diffraction (XRD), Fourier Transform-Infrared Spectroscopy (FT-IR) and UV-vis absorption spectroscopy were used to characterize the resultant modified electrodes. The properties of the electrode enable a high MP11 loading to be achieved in a stable and functional way allowing the direct reduction and oxidation of the immobilized undeca-peptide. The MP11 modified electrodes exhibited significant catalytic activity for the electrochemical reduction of ART around −0.43 V vs. Ag/AgCl in NaH2PO4 pH 7 buffer by using cyclic voltammetry (CV) and differential pulse voltammetry (DPV). The results showed a sensitive response; the cathodic catalytic current was linearly proportional to the concentration of ART in the range 0–150 μM (R = 0.9895) when using DPV with a limit of detection of 17 μM. This type of sensor has demonstrated good repeatability, reproducibility and stability and was found to be applicable for use in determining ART concentrations in extracts from the plant Artemisia annua purchased from local pharmaceutical stores. Finally due to the optical transparency of the SnO2 film electrodes, the reaction of immobilized MP11 with ART was also monitored using UV-vis spectroscopy indicating the formation of MP11/ART adducts on the surface of the film electrode.
1. Introduction
Malaria is a widespread, infectious disease, caused by a mosquito borne parasite, which continues to be a major health problem, especially in tropical and subtropical countries.1–3 More than 3 billion people suffer from the risk of malaria infection, 500 million people are infected each year, over 100 million people are vectors of this virus and up to 2.7 million consequently die each year.1,4 Therefore, research into new and superior anti-malarial drugs is necessary to gradually reduce the risk of mortality caused by the malaria parasite and control this infectious disease.Artemisinin (ART), whose discovery was part of the 2015 Nobel Prize for Medicine, is a sesquiterpene-lactone endoperoxide, isolated from the Chinese medicinal herb Quing Hao (Artemisia annua) and has successfully been used in the treatment against the resistant strains of Plasmodium falciparum for more than two decades with negligible toxicity.5–7
The peroxide group, which is part of its structure, is essential for its antimalarial activity, as its absence leads to complete loss of activity of the drug.8 It's mechanism of action involves the endo-peroxide bond of ART which breaks down by intraparasitic hemin which is produced on proteolysis of hemoglobin (Hb). Due to the breakage of the bond, free radicals are produced that kill the parasites. Therefore, there is continuing interest in developing sensitive and fast methods for studying the drug, determining its concentration and mechanism of action. However, ART is thermally unstable, stains poorly because of the absence of chromophoric and fluorophoric groups and is very sensitive to acid and base treatment. Due to these properties and behavior, its analysis and quantification by current analytical methods like Gas Chromatography (GC),9 High-Performance Liquid Chromatography (HPLC),10 as well as spectrophotometry11 and Electrospray Ionization Mass Spectrometry (ESI-MS/HPLC),12 is hindered. In addition these methods rely on complex and expensive instrumentation, require specialized personnel and are time consuming.
Furthermore, as ART contains an electrochemically active endoperoxide group, various electrochemical techniques and electrodes have been utilized for the detection of ART such as cyclic voltammetry (CV),1,13–19 square wave voltammetry,20 differential pulse voltammetry (DPV)8,16 and amperometry.2 However, some of the electrodes that were initially used for the determination of ART, such as a dropping mercury electrode, were sensitive but mercury is toxic.21 Alternatively, by using unmodified electrodes, such as glassy carbon electrode, high peak potentials were produced that made difficult the quantification of ART.21 Therefore, various materials and molecules have been used to produce modified electrodes that offer the possibility of lowering the overpotential, improving electron transfer characteristics and being more selective for the determination of ART. The electrodes that have been reported for the determination of ART include gold and glassy carbon,22 GCE electrodes,20 Hb/carbon nanofibers,19 GrO/PANI/HRP,23 MI-PDDA-RGO/GCE,24 Au-nanoparticles-chitosan modified electrodes,25 polyhydroxyalkanoate-Au/ITO,26 molecular imprinted polymers27 and Hemin modified carbon electrodes.8
The electrochemical reduction of ART is an irreversible process as ART undergoes heterolytic cleavage of its O–O bond (Fig. 1).8,19,20 Hemin (iron protoporphyrin X) has been used as a modifier on many electrodes in order to catalyze this bond cleavage through an electron transfer process, known from the interaction of ART with intraparasitic hemin of hemoglobin. MP11 is a heme undecapeptide prepared by proteolytic digestion of horse heart cytochrome c (Cyt-c) that retains the sequence Val11-Glu21 of the starting protein. The heme group of MP11 is attached covalently to the peptide chain through thioether linkages.28 The reductive activation of ART by the heme cofactor of MP11 is similar to the one described previously for hemin and hemoglobin due to the common active center (heme) of all molecules.8,19,20
 | ||
| Fig. 1 Schematic representation of MP11 immobilized on SnO2 film electrodes causing the electrocatalytic reduction of artemisinin. | ||
In this work, mesoporous SnO2/ITO film electrodes will be used for the immobilization of MP11 as they combine high surface area and porosity, optical transparency, non-toxicity, ease of fabrication and good electrochemical activity.29 These properties are important for the immobilization of enzymes, such as MP11, in a stable and functional way. In the past we managed to immobilize Cyt-c, Hb and MP11 successfully and in high amounts on the surface of these films and the immobilized molecules retained their redox and electrocatalytic activity.29,30 In this work we will for the first time use immobilized MP11 to explore the catalytic reduction of ART using CV and DPV (Fig. 1). This is the first report of MP11 immobilized on an electrode for the determination of ART, to the best of our knowledge. The well defined redox chemistry exhibited by immobilized MP11 was used to probe ART catalysis. The electrode is simple in its fabrication, exhibited good sensitivity, a low limit of detection for the determination of ART and was applicable to commercially available extracts from the plant Artemisia annua. Finally, the optical transparency of the film electrodes allows for the first time an initial study of the reaction of immobilized MP11 with ART on the surface of an electrode, using UV-vis spectroscopy and indicating the formation of MP11/ART adducts on the surface of the electrode.
2. Experimental
2.1 Chemicals
Tin(IV) oxide, 15% in H2O colloidal dispersion was purchased from Alfa Aesar. Didodecyldimethylammonium bromide (abbr. DDAB, 99.9%), absolute ethanol (99.9%), glacial acetic acid, ethyl cellulose 5–15 mPa, ethyl cellulose 30–50 mPa, terpineol, microperoxidase sodium salt (abbr. MP-11, 99.9%) and sodium dihydrogen orthophosphate (NaH2PO4) were obtained from Sigma Aldrich and used as such with no further purification. Artemisinin (abbr. ART, ≥99%) was purchased from Tocris Bioscience. Indium Tin Oxide (ITO) glass slides, of sheet resistance 15 ohm per sqr, were purchased from PsiOTec, UK.2.2 Preparation of SnO2 paste
The SnO2 paste, consisting of 15 nm-sized particles, was prepared from a 15% wt water content tin(IV) oxide colloidal solution mixed with 3 g of glacial acetic acid under ambient conditions and stirred for 36 hours in a covered Teflon flask at room temperature (RT, 25 °C). The flask was autoclaved at 240 °C for 3 days and then cooled down at RT for 5 hours. The colloids were redispersed with a 60 s cycle burst from an LDU Soniprobe horn using full power. Finally, it was triply centrifuged to remove the acid and washed with ethanol three times to produce a light green precipitate containing 40% wt SnO2 in ethanol and only trace amounts of water. After this, the colloidal solution was prepared for spreading on conducting ITO glass slides. We used a 10% SnO2 paste mixed with 28% ethyl cellulose 5–15 mPa, 22% ethyl cellulose 30–50 mPa, and 40% terpineol and ethanol as the solvent. The solution was stirred and sonicated in a JENCONS-PLS sonicator until it became homogeneous. The ethanol was removed with the use of a rotor evaporator using a vacuum pump at 40 °C.2.3 Preparation of MP-11/DDAB/SnO2/ITO electrodes
The SnO2 suspension was applied to the surface, of thoroughly cleaned in ethanol and dried, conducting ITO glass slides using a conventional “doctor blade” technique. Masking the glass slide with Scotch Magic tape, type 810, thickness 62.5 μm, provided by 3 M, controlled the thickness and the width of the area spread, with two layers of tape being employed to yield a final film thickness of ∼4 μm. The spread suspension was then allowed to dry for 15 minutes before being sintered for 30 min at 450 °C using a Nabertherm furnace. The heat burned any organic material present and sintered the SnO2 particles together providing good electrical contact throughout the film. The SnO2 glass slides were then broken up into 10 mm wide strips. In order for the films to be modified with MP-11, each 1 cm2 SnO2 film was first immersed in 20 μM DDAB, a polycationic binding promoter, overnight. The DDAB/SnO2 electrodes were then rinsed with 10 mM NaH2PO4 buffer and immersed in 15 μM MP11 solution at 4 °C overnight.2.4 Characterization of SnO2 film electrodes
The crystal structure of the SnO2 films was studied using X-ray diffraction (XRD). The XRD patterns of the SnO2 films on FTO glass were recorded using a Bruker D8 advance X-ray diffractometer with Cu Kα-radiation from 20° to 80° at a scanning speed of 0.015 deg per s. The X-ray tube voltage and current were set at 45 kV and 40 mA, respectively. The diffraction patterns were indexed by comparison with the Joint Committee on Powder Diffraction Standards (JCPDS) file 41-1445 of SnO2 cassiterite. The morphology and thickness of the SnO2 film electrodes were analysed by field emission scanning electron microscopy (FE-SEM) using a FEI inspect microscope operating at a voltage 25 kV. The specimens (films) were prepared by Au sputtering to increase the conductivity of the samples. Energy dispersive spectroscopy (EDS) was also used for the elemental analysis of the SnO2/ITO films. Fourier transform infrared (FTIR) spectroscopic analysis was carried out with a Digilab Excalibur FTS 3000MX spectrometer. The UV-visible absorption spectra were recorded using a Shimadzu UV-1800 spectrophotometer.2.5 Electrochemical/electrocatalytic measurements
Electrochemical experiments were performed using an Autolab PGStat101 potentiostat. The electrochemical cell was a three-electrode stirring cell, employing a platinum mesh flag as the counter electrode, an Ag/AgCl/KClsat reference electrode and the SnO2 film as the working electrode. All potentials are reported against Ag/AgCl. The electrolyte solution was 10 mM NaH2PO4, pH 7, which was deoxygenated with argon prior to any measurements and an argon atmosphere was kept throughout the measurements. All experiments were carried out at RT.The same setup and electrolyte were also used for the electrochemical determination of ART. An ART stock solution of 0.02 M in ethanol was prepared for the CV and DPV measurements. A microsyringe was used for injecting ART in the electrochemical cell. CVs and DPVs were taken 2 minutes after each ART addition. The DPV measurements took place in a potential range between −1.2 and 0.05 V. The optimized parameters of DPV correspond to a step potential at 5 mV, amplitude of 50 mV, modulation time of 25 ms with scan rate 100 mV s−1 and a frequency of 50 Hz. In order to determine ART concentration in real samples, the commercial ART extract was centrifuged for 10 min at 1500 rpm and then the collected sediment (5 mg ART) was dissolved in 0.9 ml ethanol, thus, achieving a concentration of 0.02 M ART. The real sample detection was tested by making three additions of 10 μL of the aforementioned solution in the electrochemical cell.
3. Results and discussion
3.1 SEM
The surface morphology and thickness of the SnO2/ITO glass film electrode was analyzed by FE-SEM. The top view FE-SEM image presented in Fig. S1a† showed that SnO2 film comprises of an inflexible, porous network of SnO2 nanoparticles of average size 20–70 nm that are evenly distributed, creating a rich mesoporous surface area. According to Fig. S1b† the thickness of the SnO2 film is around ∼4 μm as set by the adhesive tape used. Fig. S1c† displays the EDS for a SnO2/ITO film electrode carried out during the FE-SEM analysis, which comforts to the characteristic peaks of Sn and O. These results confirm that the structure of SnO2 could provide an excellent matrix for the small molecules of MP11 to diffuse throughout the ‘spongy’ porous mesostructure and provide many active sites for catalytic reactions.3.2 X-ray diffraction
The crystal structure of the SnO2/ITO glass film electrode was analysed by the XRD technique. In Fig. 2b the XRD data of the SnO2 film on ITO glass revealed peaks at 26.55°, 33.82°, 37.75° and 51.76°, corresponding to the (110), (101), (200) and (211) planes, respectively. These peaks are characteristic of cassiterite type of tetragonal rutile nanocrystals and are consistent with the reported values of the relevant JCPDS card no: 41-1445 (Fig. 2a). | ||
| Fig. 2 XRD patterns of (a) JCPDS card # 41-1445 SnO2 cassiterite and (b) SnO2/ITO glass. | ||
3.3 Fourier transform infrared analysis
FTIR spectroscopy of a protein provides information on the structure and environment of its backbone and of the amino acid chains. The amide I and II infrared bands have been used widely for monitoring conformational changes in protein structure.31Fig. 3 compares the FTIR of SnO2 and MP11/SnO2 films recorded in KBr matrices. Blank KBr pellet was taken as the background. Both spectra displayed a band at 3427 cm−1 due to O–H vibrations of adsorbed water and a band located at around 600 cm−1 due to the EU mode of SnO2 (anti-symmetric O–Sn–O).32 In comparison, the FTIR spectra of MP11/SnO2 displayed the representative absorption peaks of MP11 besides the absorption bands of SnO2. As shown in Fig. 3, we can locate new absorption bands, a large one at 1648 cm−1 assigned to amide I stretch and two smaller ones at 1450 cm−1 and 1300 cm−1 due to amide II and III respectively.33 Absorbance from DDAB does not occur in the amide region like previous studies showed in the past.33 Therefore, the coupling of MP11 on the SnO2 mesopores of our thin film was successful.
 | ||
| Fig. 3 FTIR spectra of SnO2 (black) and MP11/SnO2 (red). | ||
3.4 MP11 immobilization
Immobilization of MP11 results in orange-brown coloration of the SnO2 film electrodes. DDAB pre-modification of the surface of the films was necessary to promote MP11 immobilization, as without the use of this promoter, no significant amount of MP11 was observed on the film surface. The UV-vis absorption spectra of the MP11 on the DDAB modified SnO2 film (Fig. 4c) and of a blank SnO2 film (Fig. 4b) are presented in comparison with the respective spectra of MP11 in solution (Fig. 4a). The characteristic 405 nm band observed for MP11 in solution is red shifted to 409 nm upon immobilization on the DDAB modified SnO2 film electrode. This same observation was also reported in the past following binding of MP11 to DDAB and poly-L-lysine (PLL).30,34 This shift indicates that the heme microenvironment in immobilized MP11 is distinctly different from that in solution, indicative of the formation of low spin iron centre in the presence of these polycations.30,34 It should be noted that the blank SnO2/ITO glass film is optically transparent for wavelengths in the visible region of the spectrum, thus, it does not present any absorption peaks (Fig. 4b) and does not interfere with the absorption spectrum of MP11. Assuming that the absorption strength of MP11 is similar in the immobilized and dissolved state, an extinction coefficient of 176![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1 was employed to estimate the amount of MP11 adsorbed on the surface of the film electrode.35 This resulted in an approximate MP11 loading of 4.85 nmol on a 4 μm thick DDAB-SnO2 film electrode.
000 M−1 cm−1 was employed to estimate the amount of MP11 adsorbed on the surface of the film electrode.35 This resulted in an approximate MP11 loading of 4.85 nmol on a 4 μm thick DDAB-SnO2 film electrode.
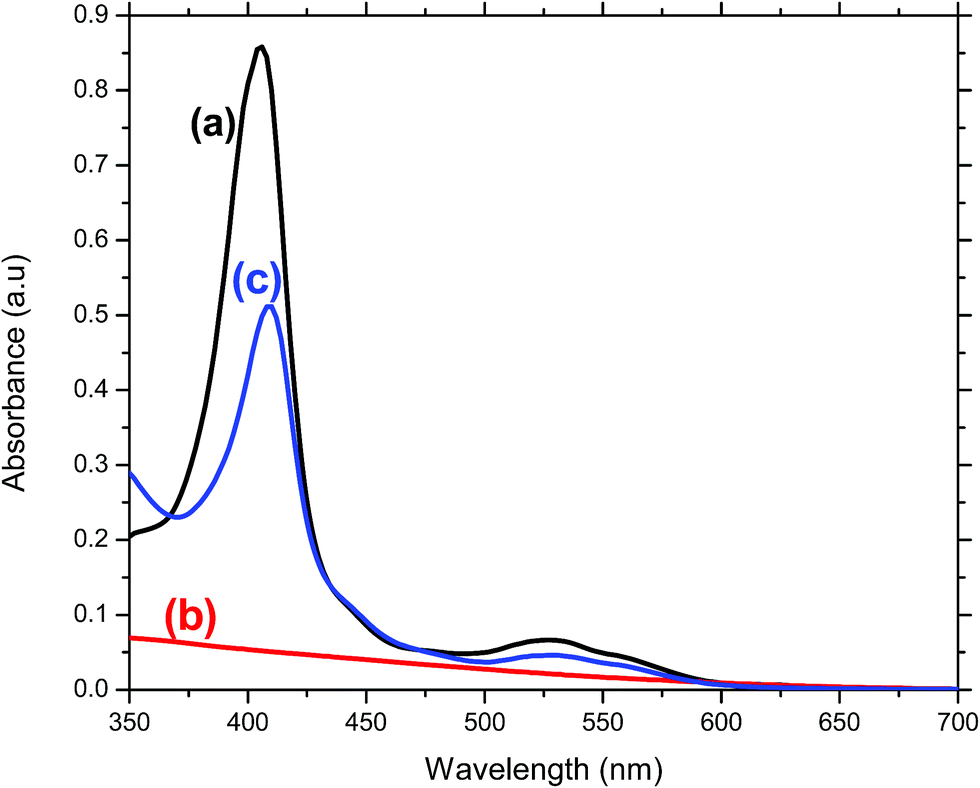 | ||
| Fig. 4 UV-vis absorbance of (a) MP11 dissolved in 10 mM NaH2PO4 pH7 buffer, (b) SnO2/ITO glass film and (c) MP11/DDAB/SnO2/ITO glass film. | ||
3.5 Electrochemical behavior of MP11/DDAB/SnO2/ITO glass electrodes
Having immobilized the MP11 on the mesoporous SnO2 electrodes successfully, the ability of the enzyme to retain its redox function after immobilization was examined. Electrochemical characteristics of the modified SnO2 electrodes were investigated by CV and all experiments were carried out in an enzyme-free, anaerobic 10 mM NaH2PO4 pH 7 buffer. Fig. 5a and b show the CVs of a ∼4 μm thick SnO2/ITO glass electrode before and after the immobilization of MP11 at different scan rates (0.01 to 0.1 V s−1). The CV of the conducting ITO glass substrate alone exhibited negligible currents. Fig. 5a shows the characteristic charging/decharging currents assigned to electron injection into sub-band gap/conduction band states of the SnO2 film and no cathodic or anodic peaks are observed, even at the slowest scan rate (0.01 V s−1). In addition, the slower the scan rate applied, the smaller the resulting current obtained. The charging of the SnO2 film starts at +0.1 V, similar to what some of the authors observed in the past for mesoporous SnO2 films sintered on the surface of other conducting glass substrates.29,30 | ||
| Fig. 5 CVs of (a) SnO2/ITO film electrode, (b) MP11/DDAB/SnO2/ITO film electrode in 10 mM NaH2PO4 (pH 7) at different scan rates and (c) plot of redox peak currents vs. scan rates. | ||
Fig. 5b shows that after the immobilization of MP11 on the films surface, a couple of redox peaks are observed, a cathodic and an anodic peak at −0.43 V and −0.2 V (vs. Ag/AgCl) respectively (acquired at 0.1 V s−1), attributed to heme reduction and oxidation. A current increase is clearly observed as scan rates are steadily increased, a clear indication of the involvement of surface confined electroactive species (MP11). The midpoint redox potential of immobilized MP11 was determined to be −0.32 V, very close within error to previous studies of MP11 on very similar electrodes30 and close to the midpoint potential of MP11 in solution which is −0.377 V.36 The midpoint potential of the immobilized MP11 is shifted to a more positive value due to the presence of the binding promoter DDAB on the films surface which is expected to provide a positive and hydrophobic microenvironment for the immobilized MP11. Positive microenvironments may be expected to increase the electron affinity of a redox couple, hence inducing a positive shift of the enzymes midpoint potential and at the same time the hydrophobic environment stabilizes the heme porphyrin of the immobilized MP11.30 A similar observation was also reported by Huang when studying MP11 immobilized on DDAB modified PEG electrodes34 and by Astuti on very similar, as in this study, SnO2 electrodes.30 Rusling, also reported in the past a positive shift of the midpoint potential of several proteins immobilized on electrodes modified with this cationic lipid film.37
The peak current area of the cathodic peak measured in the CV (Fig. 5b) corresponds to the amount of charge transferred to the electrode and therefore the amount of adsorbed MP11 that participates in the interfacial electron transfer. For a scan rate of 0.05 V s−1, the amount of immobilized MP11 is estimated to be ∼5 nmol. Comparison of this data with the spectroscopy data of immobilized MP11 (4.85 nmol) gives estimation that all immobilized protein is electroactive. The amount of MP11 immobilized on the surface of the film electrode was also estimated by measuring the absorbance of MP11 solution before and after the immersion of the DDAB/SnO2/ITO film electrode in it and further confirmed that the amount of immobilized MP11 is ∼5 nmol.
Fig. 5c shows the plot of current vs. scan rate for a MP11/DDAB/SnO2/ITO glass film, displaying a linear correlation for scan rates up to 0.1 V s−1. This allows the use of Laviron model to analyze the kinetics of heterogeneous electron transfer.38 The peak to peak potential separation (ΔEp) is 110 mV at a scan rate of 0.05 V s−1, similar to other MP11 modified electrodes reported in the past.30,39 This implies a relatively fast direct electron transfer between the redox active center of heme of MP11 and the modified film electrode. A graph of the dependence of peak separation on the logarithm of the scan rate yields a straight line with a slope equal to charge transfer coefficient, α, of 0.08. For a peak separation 110 mV and a scan rate 0.05 V s−1, an interfacial electron transfer rate constant, ks, value of MP11 on the modified SnO2 films was estimated to be 0.3 ± 0.05 s−1. This value is in the range of ks for typical surface quasi-reversible electron transfer.
3.6 Electrochemical ART biosensor
Having immobilized MP11 successfully on the DDAB modified SnO2 film electrodes in an electroactive configuration, its electrocatalytic activity toward ART reduction is examined. Fig. 6 shows the CVs of a MP11/DDAB/SnO2/ITO glass film after the addition of increasing concentrations of ART (0–200 μM) at 0.05 V s−1 scan rate. ART has a very low solubility in water and hence it was dissolved in ethanol where its solubility is improved as reported previously by other groups.19 The reduction and oxidation peaks of the immobilized MP11 were visible as before, only this time the addition of ART caused a gradual increase of the cathodic (reduction) current and a gradual small decrease of the anodic oxidation current. Firstly, the immobilized MP11 molecules with their hemin electroactive center, are reduced from the MP11(Fe3+) to MP11(Fe2+). After the addition of ART, the molecules of reduced MP11 catalyse the peroxidase bond between the oxygens in the aromatic ring of ART. When this bond has been broken, the molecules of ART are reduced and the MP11(Fe2+) is re-oxidised to MP11(Fe3+). The cathodic reduction current increases in correlation with the increasing concentration of ART. The details of the proposed mechanism are given below:MP11(Fe3+) + H+ + e− ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) MP11(Fe2+) MP11(Fe2+) |
| ART–(O–O)– + MP11(Fe2+) → ART–(O−)2 + MP11(Fe3+) |
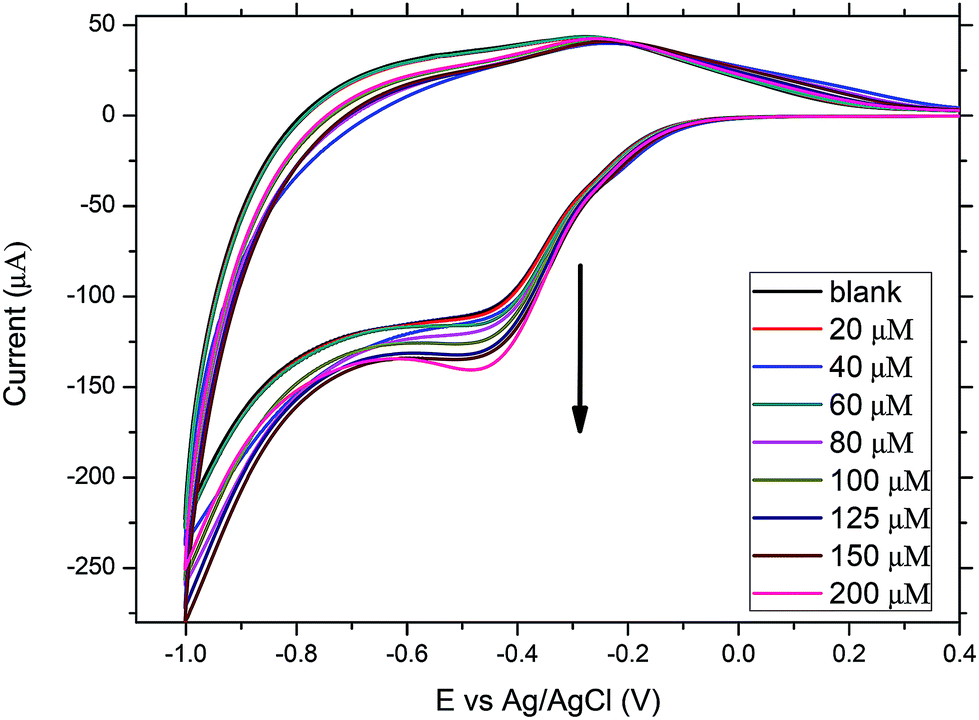 | ||
| Fig. 6 CVs of MP11/DDAB/SnO2/ITO film electrode at 0.05 V s−1 in the absence and presence of increasing concentrations of ART. | ||
There is indication that the interaction of ART with the immobilized MP11 is a diffusion controlled process, as for scan rates faster than 0.1 V s−1, the changes in the cathodic and anodic peaks are less clear, as there is not enough time for ART to diffuse to the electrode surface for its reduction to occur. Usually, in diffusion controlled processes, catalytic currents are only observed at low scan rates.19 The proposed mechanism of reaction is similar to the one proposed by Mazzochette for the electrochemical catalysis of ART on haemoglobin functionalized carbon nanofibers.19 The influence of the microenvironment provided by DDAB did not affect the catalytic activity of MP-11. This observation was obtained regardless of the type of binding promoters used as we monitored similar behavior when we used poly-L-lysine (PLL) instead of DDAB. The reason we used DDAB and not PLL was that we managed to immobilize twice the amount of MP11 on the films surface when using DDAB.
DPV is more sensitive than CV and therefore has been extensively used as a more sensitive method for the detection of low concentration of analytes. The DPV parameters that were used for the detection of ART, were found to correspond to a modulation amplitude of 50 mV, a step potential of 5 mV and a modulation time of 25 ms. The potential applied to the MP11/DDAB/SnO2/ITO film electrode was between −0.6 V and 0 V at a scan rate of 0.1 V s−1. Fig. 7a depicts the differential pulse voltammograms (DPVs) recorded for different ART concentrations (0–350 μM). The cathodic peak current at −0.44 V of MP11 increases with increasing concentrations of ART added in the 10 mM NaH2PO4 pH 7 electrolyte solution, as explained in the mechanism presented before.
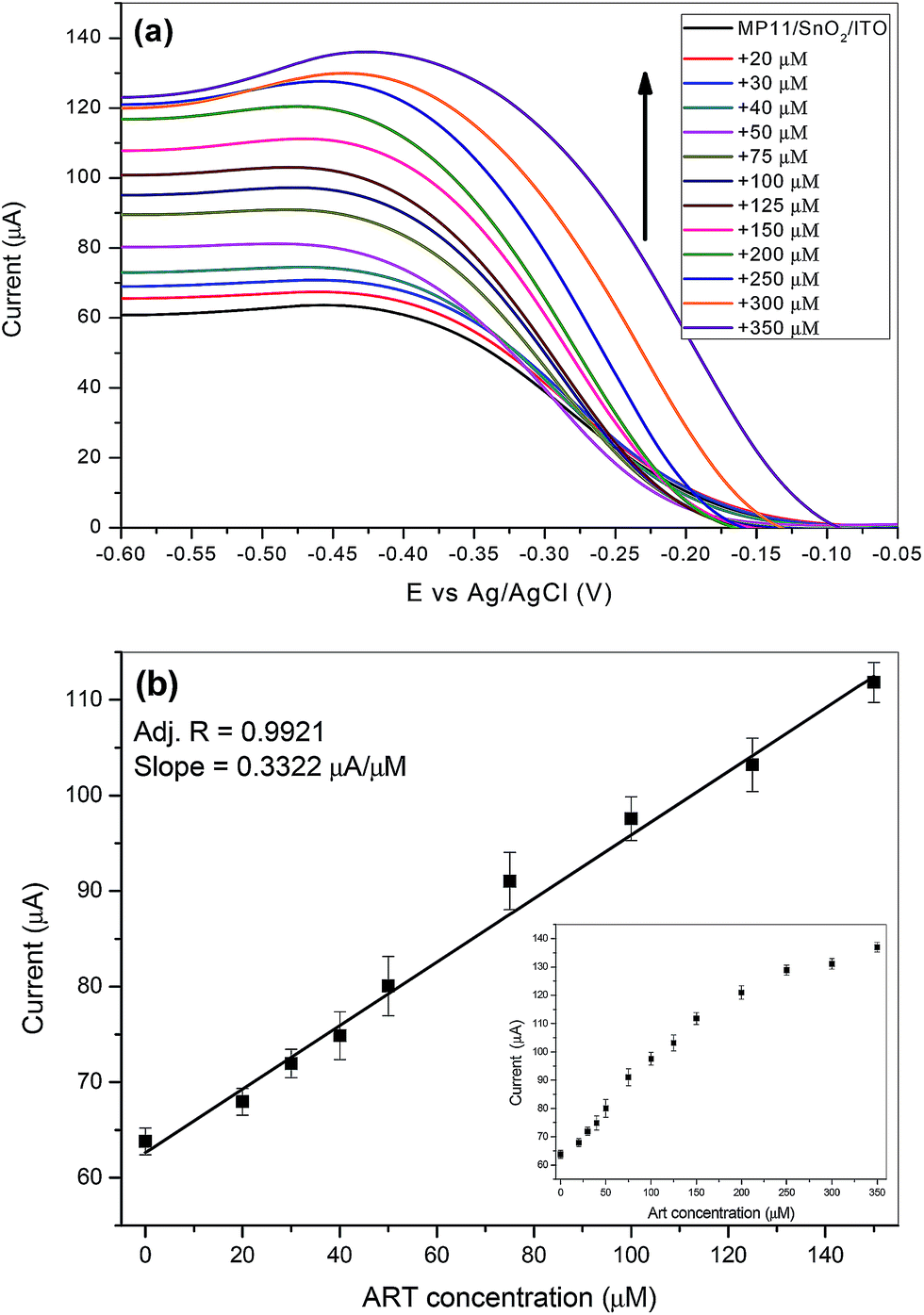 | ||
| Fig. 7 (a) DPVs of a MP11/DDAB/SnO2/ITO film electrode after the addition of increasing ART concentrations in 10 mM NaH2PO4 pH 7 buffer at a scan rate 0.1 V s−1 and (b) calibration plot between the anodic peak currents and ART concentration. | ||
It was found that the peak currents were linear to the ART concentration for the range from 0–150 μM with a good correlation coefficient of 0.9921 as shown in Fig. 7b. The limit of detection (LOD) of the ART biosensor was calculated to be 17 μM from the slope of the calibration plot and based on a signal-to-noise ratio of 3.
The analytical performance of our MP11/DDAB/SnO2/ITO film electrode towards ART determination is compared with other modified electrodes for ART detection, using the same or other electrochemical techniques and is presented in Table 1. It is apparent that the proposed sensor's sensitivity (linear range) and LOD are in the μM range, comparable to most of the previously presented electrochemical ART sensors. However, some modified electrodes exhibit LOD in the nM range. The lower sensitivity of our sensor is not because MP11 is not properly oriented to facilitate electron transfer reactions, neither that our electrode does not provide a large surface area for the immobilization MP11 and therefore many catalytic sites for the reduction of ART. Our results indicate that it could be the relatively large volume of our electrochemical cell which is 10 ml, most of the cells used for other modified electrodes mentioned in Table 1 are 5 ml or less. However, in this work, we would like to present a simple, low cost sensor for the determination of ART. Almost all other electrodes displayed on Table 1 are of considerably higher production cost and require more complicated and lengthy preparation processes. In addition, they are not optically transparent and therefore do not allow the reaction of MP11 with ART to be monitored optically as well and examine the reaction mechanism in more detail, as we did in Section 3.8.
| Electrodes | Techniques | Linear range | LOD/Sensitivity | Reference |
|---|---|---|---|---|
| Hemin on silica gel/TiO2 | CV/amperometry | 50–1000 nM | 15 nM | 2 |
| Cobalt phthalocyanine/carbon paste electrode | CV/DPV | 21–530 μM | 16.5 μM | 8 |
| Hb/carbon nanofibers | CV | 0–200 μM | 0.329 mA mM−1 | 19 |
| Hemin/carbon fat electrode | CV/DPV | 4.8–78 μM | 1.4 μM | 21 |
| Graphene oxide/polyaniline/HRP electrode | CV/impedance spectroscopy | 5–40 nM | 1.2–1.4 nM | 23 |
| HRP-polyhydroxyalkanoate–Au nanoparticles/ITO | CV | 10–80 nM | 3.6 nM | 26 |
| MP11/DDAB/SnO2/ITO | CV/DPV/UV-vis | 0–350 μM | 17 μM | This work |
The reproducibility of our modified electrodes was also tested by preparing 5 separate modified SnO2 film electrodes under the same conditions that gave very similar results for the determination of ART with a relative standard deviation of around 5%. Finally the stability of our ART biosensor was tested. It was found that after electrochemical cycling, very little amount of MP11 (up to 5%) got desorbed from the surface of the film. The film was then washed thoroughly with buffer and stored at 5 °C for up to 1 week before it was used again for the determination of ART. It was found that it retains at least 90% of its catalytic activity. However, a prolonged usage of these films is not recommended and in order to maintain the highest sensitivity across all new measurements, fresh samples should be used accordingly.
3.7 Analysis of real samples
The applicability and validity of the proposed MP11/DDAB/SnO2/ITO electrode was evaluated for the determination of ART in pharmaceutical extracts of the Artemisia annua plant, purchased from a local pharmaceutical store. The real sample analysis was performed using the CV and DPV technique, where the measurements were carried out using the setup and pretreatment protocol described in the Experimental section. The recovery of ART in these real samples, based on three replicate measurements is reported in Table 2. The CVs of one of these measurements is presented in Fig. S2,† where the shape, size and peaks of the CVs are similar to the ones presented in Fig. 6 for the addition of increasing concentrations of ART. The results show satisfactory recoveries of ART with the percentage in the range of 98.5% to 104%, thus, confirming the efficient applicability of the proposed electrochemical ART sensor in real sample analysis.| Sample | Spiked (μM) | Detected (μM) | Recovery (%) |
|---|---|---|---|
| 1 | 20 | 20.8 | 104 |
| 2 | 40 | 40.5 | 101.2 |
| 3 | 60 | 59.1 | 98.5 |
3.8 MP11/DDAB/SnO2/ITO film electrodes used for optical studies
The optical transparency of the MP11/DDAB/SnO2/ITO electrodes allows the investigation of the interaction of immobilized MP11 with ART using also UV-vis spectrometry and monitoring changes in the absorption spectrum. This is unusual, as most electrodes used for electrochemical sensors are not optically transparent and do not allow such study (none so far has been reported with this property for the determination of ART). MP11 was immobilized on the modified film surface as described before and was immersed in a cuvette with 10 mM NaH2PO4, pH 7 buffer, where increasing concentrations of ART (6–33 μM) were added. Fig. 8 shows the UV-visible spectra of immobilized MP11 with increasing concentrations of ART. The Soret peak at 407 nm is clearly observed due to the heme group of immobilized MP11. After the additions of ART, no new peaks were observed but the absorbance intensity of the Soret peak decreased gradually and a blue shift by 5–6 nm was monitored from 407 to 401 nm. Two literature reports in the past19,40 reported that reaction of hemoglobin (Hb) in solution (heme protein) with ART caused the decrease of the Soret peak and a blue shift of 6 nm. | ||
| Fig. 8 UV-vis absorption spectra of MP11/DDAB/SnO2/ITO glass film with increasing concentrations of ART (0–33 μM). | ||
According to Fig. 8, the interaction of immobilized MP11 with ART is at least biphasic. During the first period covering 30 min, the intensity of the Soret band is decreased by 14% and a small blue shift is observed. Afterwards, a progressive continuous decrease in intensity of the Soret band takes place, after 2 hours the absorption intensity is reduced by 40% and after 24 hours by 65%. Only the heme group of MP11 absorbs at 407 nm and the decrease or shift of the Soret band intensity is due to the interaction of ART with immobilized MP11. The heme group of immobilized MP11 is attacked and degraded by ART.
According to a recent report of Mazzochette, performing a similar study by adding increasing concentrations of ART to Hb in solution, monitoring the absorption changes and performing liquid chromatography analysis of samples of Hb incubated with ART, he concluded that many types of adducts may form between Hb and ART.19 The mechanism of action of ART likely goes through several transient radical intermediates arising from cleavage of the endoperoxide bond.15 However, in his electrochemistry experiments using Hb modified carbon nanofiber electrodes, the electrocatalytic reduction of Hb exhibited a single peak and no evidence of multiple intermediates were obtained. This is similar to the single peak we obtained for our MP11/DDAB/SnO2 electrodes due to the electrocatalytic reduction of the immobilized MP11. Although the CVs obtained were repeated numerous times, there was no significant change in size or peak position indicating that the binding of ART did not occur on the surface of the electrode and formation of adducts did not take place. The reaction of the reduced immobilized MP11 with ART on the electrode surface caused the generation of a catalytic current proportional to the concentration of ART. This is similar to other reports2,15,19 using heme containing molecules on electrodes that generate catalytic currents due to the presence of ART and is also similar to the reactions of Hb or heme with H2O2 on electrodes reported recently also by our group using SnO2 electrodes.41
4. Conclusions
This work has demonstrated that mesoporous SnO2 films on ITO glass substrates, prepared by a simple method, are an ideal support for MP11 immobilization in a stable and functional way. The MP11 is able to retain its redox activity upon immobilization without the use of electron transfer mediators and with a fast electron transfer rate constant of 0.3 ± 0.05 s−1. It was then used to investigate the behavior of ART electroreduction by exhibiting high electrocatalytic activity, generating catalytic currents proportional to the concentration of ART. In addition, it allowed the ART reduction potential to less negative values when compared to other electrodes. The modified electrode in phosphate buffer, pH 7 yielded a relatively low detection limit, good sensitivity, repeatability and stability for ART. Finally, the transparency of the SnO2 films allowed the interaction of immobilized MP11 with ART to be monitored also optically. Many adducts could have been formed as a result of the cleavage of the endoperoxide bond of ART with immobilized ART but the single reduction peak of the CVs suggests otherwise. Lastly, our modified electrode could be used successfully as an efficient catalyst for ART reduction in aqueous solutions, in potential analysis of pharmaceutical products or in field experiments.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was partially supported by Grant 80669 from the Research Committee of the University of Patras via “C. CARATHEODORI” program.References
- Y. Chen, J. M. Zheng, S. M. Zhu and H. Y. Chen, Electrochim. Acta, 1999, 44, 2345–2350 CrossRef CAS.
- J. R. M. Reys, P. R. Lima, A. G. Cioletti, A. S. Ribeiro, F. Caxico de Abreu, M. O. F. Goulart and L. T. Kubota, Talanta, 2008, 77, 909–914 CrossRef CAS.
- N. Towie, Nature, 2006, 440, 852–853 CrossRef CAS.
- World Health Organization, The World Health Report 1996-Fighting disease, Fostering Development, 1996, Geneva, Switzerland Search PubMed.
- P. Christen and J. L. Veuthey, Curr. Med. Chem., 2001, 8, 1827–1839 CrossRef CAS PubMed.
- P. J. De Vries and T. K. Dien, Drugs, 1996, 52, 818–836 CrossRef CAS PubMed.
- E. Gkranias-Klotsas and A. M. Lever, Blood Rev., 2007, 21, 73–87 CrossRef PubMed.
- C. Debnath, P. Saha and A. Ostrner, Electroanalysis, 2008, 21, 657–661 CrossRef.
- C. A. Peng, J. F. S. Ferreira and A. J. Wood, J. Chromatogr. A, 2006, 1133, 254–258 CrossRef CAS PubMed.
- N. Ermedoglu, I. Orhan, M. Kartal, N. Adiguzel and B. Bani, Rec. Nat. Prod., 2007, 1(2–3), 36–43 Search PubMed.
- J. Suberu, L. Song, S. Slade, N. Sullivan, G. Barker and A. A. Lapkin, J. Pharm. Biomed. Anal., 2013, 84, 269–277 CrossRef CAS PubMed.
- L. Messori, F. Piccioli, B. Eitler, M. C. Bergonzi, A. Bilia and F. Vincieri, Bioorg. Med. Chem. Lett., 2003, 13, 4055 CrossRef CAS PubMed.
- H. H. Cai, J. Cai and P. H. Yang, Bioorg. Med. Chem., 2009, 19, 863–866 CrossRef CAS.
- F. C. Gong, Z. D. Xiao, Z. Cao and D. X. Wu, Talanta, 2007, 72, 1453–1457 CrossRef CAS PubMed.
- P. H. Yang, Z. J. Zhou and J. Y. Cai, Colloids Surf., A, 2005, 257–258, 467–472 CrossRef CAS.
- H. Bai, C. Wang, J. Chen, J. Peng and Q. Cao, Biosens. Bioelectron., 2015, 64, 352–358 CrossRef CAS PubMed.
- R. L. Donkers and M. S. Workentin, J. Phys. Chem. B, 1998, 102, 4061–4063 CrossRef CAS.
- F. Zhang, D. K. Gosser and S. R. Meshnick, Biochem. Pharmacol., 1992, 43, 1805–1809 CrossRef CAS PubMed.
- Z. Mazzochette, E. Newton and A. Mugweru, Anal. Methods, 2017, 9, 2997–3002 RSC.
- R. Jain Vikas, Colloids Surf., B, 2011, 88, 729–733 CrossRef PubMed.
- C. Debnath, P. Saha and A. Ortner, Electroanalysis, 2008, 20, 1549–1555 CrossRef CAS.
- A. M. Mugweru, Int. J. Chem. Kinet., 2016, 48, 72 CrossRef CAS.
- K. Radhapyari, P. Kotoky, M. R. Das and R. Khan, Talanta, 2013, 111, 47–53 CrossRef CAS PubMed.
- J. Zhou, X. Sun and K. Wang, Int. J. Electrochem. Sci., 2016, 11, 3114–3122 CrossRef CAS.
- F. Gong, Z. D. Xiao, Z. Cao and D. X. Wu, Talanta, 2007, 72, 1453–1457 CrossRef CAS PubMed.
- P. Phukon, K. Radhapyari, B. K. Konwar and R. Khan, Mater. Sci. Eng., C, 2014, 37, 314 CrossRef CAS PubMed.
- A. F. T. Waffo, C. Yesildag, G. Caserta, S. Katz, I. Zegber, M. C. Lensen, U. Wollenberger, F. W. Scheller and Z. Altintas, Sens. Actuators, B, 2018, 275, 163–173 CrossRef CAS.
- M. Rodriguez, C. Claparols, A. Robert and B. Meunier, ChemBioChem, 2002, 11, 1147–1149 CrossRef.
- E. Topoglidis, Y. Astuti, F. Duriaux, M. Gratzel and J. R. Durrant, Langmuir, 2003, 19, 6894–6900 CrossRef CAS.
- Y. Astuti, E. Topoglidis and J. R. Durrant, Anal. Chim. Acta, 2011, 686, 126–132 CrossRef CAS PubMed.
- T. F. Kumosinski and J. J. Unruh, Molecular Modeling, ed. T. F. Kumosinski and M. M. Leibman, ACS Symposium Series, Washington, DC, 1993, vol. 576, pp. 71–98 Search PubMed.
- H. Seema, K. C. Kemp, V. Chandra and K. S. Kim, Nanotechnology, 2012, 23, 355705 CrossRef PubMed.
- W. Huang, J. Jia, Z. Zhang, X. Han, J. Tang, J. Wang, S. Dong and E. Wang, Biosens. Bioelectron., 2003, 18, 1225–1230 CrossRef CAS PubMed.
- W. Huang, Z. Zhang, X. Han, J. Tang, Z. Peng, S. Dong and E. Wang, Biophys. Chem., 2001, 94, 165–173 CrossRef CAS PubMed.
- T. Tatsuma and T. Watanabe, Anal. Chem., 1991, 63, 1580–1585 CrossRef CAS PubMed.
- L. Gorton, A. Lindgren, T. Larsson, F. D. Munteanu, T. Ruzgas and I. Gazaryan, Anal. Chim. Acta, 1999, 400, 91–108 CrossRef CAS.
- J. F. Rusling and A. E. F. Nassar, J. Am. Chem. Soc., 1993, 115, 11891–11897 CrossRef CAS.
- E. Laviron, J. Electroanal. Chem., 1979, 101, 19–28 CrossRef CAS.
- T. Loetzbeyer, W. Schuhmann and H. L. Schmidt, Sens. Actuators, B, 1996, 33, 50–54 CrossRef CAS.
- R. Kannan, K. Kumar, D. Sahal, S. Kukreti and V. S. Chauhan, Biochem. J., 2005, 385, 409–418 CrossRef CAS PubMed.
- A. Panagiotopoulos, A. Gkouma, A. Vassi, C. J. Johnson, A. E. G. Cass and E. Topoglidis, Electroanalysis, 2018, 30, 1956–1964 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ay00764d |
| This journal is © The Royal Society of Chemistry 2019 |