DOI:
10.1039/C9AY00573K
(Paper)
Anal. Methods, 2019,
11, 2960-2968
SERS-based sandwich bioassay protocol of miRNA-21 using Au@Ag core–shell nanoparticles and a Ag/TiO2 nanowires substrate
Received
20th March 2019
, Accepted 7th May 2019
First published on 28th May 2019
Abstract
Based on surface-enhanced Raman scattering (SERS) technology, Au@Ag@4MBA@5′-NH2-ssDNA probes and a Ag/TiO2@3′-NH2-ssDNA substrate were prepared and constructed into a sandwich structure to develop a high sensitivity bioassay of miRNA-21. The Au@Ag@4MBA@5′-NH2-ssDNA probes were prepared by immobilizing 5′-NH2-ssDNA onto the surfaces of 4MBA-labelled Au@Ag core–shell nanoparticles, and the Ag/TiO2@3′-NH2-ssDNA substrate was prepared by immobilizing 3′-NH2-ssDNA on the surface of Ag/TiO2 nanowires SERS-active substrates. The experimental results showed that the SERS-based sandwich bioassay of miRNA-21 presented a low limit of detection of 0.75 fM and a broad dynamic range from 1.0 fM to 1.0 nM. Also, the test data for the SERS-based sandwich bioassay were not only consistent with that of the real-time fluorescence quantitative polynucleotide chain reaction (RT-qPCR) method but also displayed higher detection sensitivity. It was shown that the SERS-based sandwich bioassay of miRNA-21 has importance for use in potential applications involving diagnosing clinical cancer patients.
1. Introduction
To date, cancer represents one of the most serious threats to human health and life due to the high morbidity and mortality of cancer patients.1 The latest version of the World Cancer Report (2014) predicted that global cancer cases would show a rapid growth from 14 million in 2012 to 19 million in 2025 and up to 24 million by 2035, with lung cancer being the most common and fatal cancer, with about 1.8 million new cases annually and 1.59 million deaths in 2012, with China accounting for more than one-third of such cases.2 Faced with such a very serious situation, there is an increasingly urgent requirement for the better diagnosis and treatment of cancer.
In recent decades, a great diversity of medical methods have been developed to diagnosis cancer. Currently, cytological examination, imaging technology, biopsy and tumour marker assays are the most popular methods used to screen and diagnose cancer. In general, cytological examination is only used to qualitatively diagnose tumours by observing the histopathological changes of cells. Regarding imaging technology, including computed tomography (CT), X-rays and magnetic resonance imaging (MRI), this is useful as it can provide a visual and high-resolution imaging of organic tissues, but an accurate diagnosis is also dependent on the experience and skill of the doctor.3–5 Although the biopsy method can provide an effective and accurate histopathologic diagnosis, a frustrating thing is that the patients have to endure the pain of surgery.6 Furthermore, even if the above methods can make a definite diagnosis, most patients will already be in the middle or terminal stages of cancer, which makes it difficult to cure them. Fortunately, the tumour marker assay has been developed into an effective and high throughput analysis method for the early screening of cancer due to the close correlation between cancer and the tumour marker. For example, much research has shown that, as typical protein tumour markers, prostate specific antigen (PSA), alpha-fetoprotein (AFP) and carbohydrate antigen (CA19-9) are strongly associated with prostate cancer, liver cancer and pancreatic cancer, respectively.7–9 Also, the enzyme-linked immunosorbent assay (ELISA), the fluorescence-based immunoassay (FBIA) and the chemiluminescence enzyme immunoassay (CLIA) have been successfully applied to high-sensitivity assays of protein tumour markers.10–12 In addition, it has been revealed that microRNAs (miRNAs) have a significant biological function to regulate the developmental and physiological processes of the human body, and consequently, various technologies have been developed for the quantitative analysis of the dynamic expression of miRNA tumour markers.13–15 For example, Micheal et al. analysed the expression of miRNA sequences in colon cancer and normal mucosa by Northern blotting and found that miRNA-143 and miRNA-145 were significantly lower in tumour tissues than normal tissues.16 Wen et al. explored the different expression levels of miR-486-5p in papillary thyroid carcinoma (PTC) and normal thyroid samples based on microarray analysis and miRNA sequencing and demonstrated that the downregulation of miR-486-5p expression was associated with PTC.17 Interestingly, owing to the fact that miRNA-21 plays an important role in the occurrence and development of many diseases, such as cancers, cardiovascular and pulmonary diseases, Huang's group proposed two effective protocols for the ultrasensitive detection of miRNA-21, which were respectively based on the signal amplification of tungsten oxide-graphene composites coupled with a target-catalyzed hairpin assembly as well as competitive RNA–RNA hybridization immobilized on the surface of tungsten diselenide nanosheets.18,19 Markou et al. used the real-time fluorescence quantitative polynucleotide chain reaction (RT-qPCR) method to identify the expression profile of mature miRNA-21 and mature miRNA-205 in non-small cell lung cancer (NSCLC) and their surrounding normal tissues, which indicated the overexpression of mature miRNA-21 and mature miRNA-205 in lung cancer tissues.20 Although the applications of the above methodologies display several advantages, such as high throughput, good reliability and good quantitative capacity, they are very expensive, time-consuming and unsuitable for in situ clinical analysis. Therefore, it is expected that the high-sensitivity and specificity trace analysis of miRNA-21 could help realize the diagnosis of NSCLC in the early stages.
On the other hand, as a non-destructive approach, the surface-enhanced Raman scattering (SERS) effect of noble metal nanostructures can greatly enhance the Raman signal of adsorbed molecules on a metal surface and could be applied to biological and medical detection due to the highly sensitive, fast and unique fingerprint characteristics of such an approach.21–23 As is well known, the excellent SERS characteristic of IB group elements-based nanostructures originates from their unique localized surface plasmon resonance (LSPR) properties, which not only depend on their structure but also on their components, such as gold or silver.24,25 Generally, the SERS enhancement effect of Ag nanoparticles (NPs) is better than that of Au NPs, while Au NPs can be easily synthesized with uniform particle size control and stable performance. In particular, silver shell and gold core NPs (Au@Ag core–shell NPs) have excellent properties from the two kinds of single precious metals, namely Au and Ag, combining the good homogeneity of gold with the high enhancement factor of silver.26 Recently, Yang's group proposed a new SERS analysis strategy combining a stable SERS reporter element and duplex-specific nuclease (DSN)-assisted signal amplification for the quantitative detection of exosomal miRNA extracted from human blood.27 By using silica-coated, analyte-tagged gold nanoparticles (SA@GNPs) and gold-coated paramagnetic nanoparticles (Au@MNPs), Zhang et al. developed a SERS-based sandwich bioassay to quantitatively and reproducibly detect miRNA-14.28 Also, Pang et al. designed a functionalized Fe3O4@Ag magnetic nanoparticle (NP) biosensor for the ultrasensitive detection of miRNA let-7b with an extra low detection limit of 0.3 fM.29 As can be seen from the above, SERS-based bioassay strategies have demonstrated superiority for detecting specific miRNAs, but still face many challenges for the accurate quantitative of miRNAs because of the low miRNA content within peripheral blood and due to the easy degradability of miRNA after the sampling of serum.30 Actually, how to raise the detection sensitivity also relies on the SERS-active substrates having a better performance. For decades, a great deal of attention has been paid to fabricate SERS-active substrates with both a high density of active spots and a homogenous surface by extending work from precious metal to non-IB group materials, such as transition metals, metal oxides semiconductors and metal halides.31 Typically, various gold- or silver-decorated TiO2 hybrid nanostructures have been proven to be excellent SERS-active substrates, where their SERS enhancement results from the electromagnetic enhancement of the noble metal NPs and the chemical enhancement of the metal oxides.32,33 Thus, besides a good synthesis of noble metal NPs, it would seem reasonable to prepare a stable and reproducible SERS-active substrate by decorating gold or silver on semiconductors with special structures for satisfying the detection requirement of trace miRNA.
In this work, based on the principle of the complementary base pairing of DNA, we propose a SERS-based sandwich bioassay protocol for implementing the ultrasensitive detection of a target analyte. First, 5′-NH2-ssDNA was linked with 4-mercaptobenzoic acid (4MBA)-covered Au@Ag core–shell NPs to synthesize Au@Ag@4MBA@5′-NH2-ssDNA probes. Also, 3′-NH2-ssDNA was immobilized on the silver NPs-decorated titanium dioxide nanowire (Ag/TiO2 NWs) substrate modified with 11-mercaptoundecanoic acid (11-MUA) and 11-sulfanylundecanol (11-MU) to obtain the Ag/TiO2@3′-NH2-ssDNA substrate. Then, the Au@Ag@4MBA@5′-NH2-ssDNA probes and the Ag/TiO2@3′-NH2-ssDNA substrate separately formed hybrids with miRNA-21 to construct a sandwich structure as the bioassay platform of miRNA-21. The experimental results showed that the quantitative detection of miRNA-21 had a wide concentration range from 1.0 fM to 1.0 nM and an ultralow limit of detection (LOD) of 0.75 fM. Also, the specificity of the SERS-based sandwich bioassay protocol was checked by using non-complementary miRNA as the target miRNA. Moreover, the SERS-based detection results were well consistent with that of the RT-qPCR method, which verified the reliability and practicability of the SERS-based sandwich bioassay method.
2. Materials and methods
2.1 Materials
Silver nitrate (AgNO3, 99.5%), tetrachloroauric acid (HAuCl4·3H2O), N-hydroxysulfosuccinimide sodium salt (NHS) and PBS/0.05% Tween buffer solution (0.05 M Tris, 0.138 M NaCl, 0.0027 M KCl, 0.05% Tween 20, pH = 8) were purchased from Sigma-Aldrich (Milwaukee, WI, USA). 4-Mercaptobenzoic acid (4MBA) was purchased from Tokyo Chemical Industry Co., Ltd (Tokyo, Japan). Sodium citrate was obtained from Tianjin Bodi Chemical Co., Ltd. (Tianjin, China). Titanium sheet (0.2 mm, 99.99%) was purchased from Shenzhen Honglei Metal Material Co., Ltd (Shengzhen, China). Ascorbic acid (AA), sodium hydroxide (NaOH), hydrofluoric acid (HF), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), 11-mercaptoundecanoic acid (11-MUA) and 11-sulfanylundecanol (11-MU) were purchased from Aladdin Chemistry Co., Ltd (Shanghai, China). Tris–EDTA buffer solution (10 × TE) was obtained from Suzhou Zeke Biotechnology Co., Ltd (Suzhou, China). All the oligonucleotides and DEPC-H2O were obtained from Generay Biotech (Shanghai) Co., Ltd (Shanghai, China). The oligonucleotides sequences are shown in Table 1. Milli-Q water (18.2 MΩ cm) was used for all the solution preparations. Glassware was cleaned by aqua regia and rinsed with deionized water prior to the experiment.
Table 1 Oligonucleotide sequences
| Oligonucleotide |
Sequence |
| miRNA-21 |
5′-UAGCUUAUCAGACUGAUGUUGA-3′ |
| Non-complementary-miRNA-106a |
5′-AAAAGUGCUUACAGUGCAGGUAG-3′ |
| Non-complementary-miRNA mimics |
5′-UUGUACUACACAAAAGUACUG-3′ |
| 5′-NH2-ssDNA |
5′-NH2-AAAAATCAACATCAIT-3′ |
| 3′-NH2-ssDNA |
5′-CTIATAAICTAAAAAA-NH2-3′ |
2.2 Synthesis of the Au@Ag@4MBA@5′-NH2-ssDNA probes
The Au@Ag core–shell NPs were prepared by the seed growth method via a two-step reaction in aqueous solution.34 The detailed steps are described as follows. First, HAuCl4 aqueous solution (100 mL, 0.01 wt%) was added to a flask and boiled with stirring. Then, sodium citrate (1.20 mL, 1 wt%) was quickly added to the boiling HAuCl4 solution and refluxed for 30 min. When the colour of the mixed solution changed from transparent to wine red, it indicated that the Au NPs had been successfully obtained and so the solution was gradually cooled to room temperature for further use. Second, seven Au NPs solutions of 10 mL were taken and mixed with AA (1.50 mL, 0.1 M) in a round flask by magnetic stirring, respectively. Next, different amounts of AgNO3 (1.0 mM) solutions (0.50, 1.0, 1.50, 2.0, 2.50, 3.0, 3.50 mL) were added dropwise to the above mixtures at a rate of 0.40 mL h−1via a syringe pump, respectively. Under magnetic stirring, Ag+ was reduced into Ag by AA and continuously grew at the surface of Au NPs. When the colour of the solutions changed gradually from wine red to orange-yellow, it indicated that Au@Ag core–shell NPs with different thicknesses of Ag shell had been synthesized. Subsequently, all the Au@Ag core–shell NPs solutions were purified by centrifugation at 8000 rpm three times for 15 min each time and the resulting products were dispersed in 3 mL of deionized water, respectively.
Furthermore, the Au@Ag@4MBA@5′-NH2-ssDNA probes were prepared as follows. First, 4MBA ethanol solutions (10 μL, 1 mM) were added into the above purified Au@Ag core–shell NPs solutions of 3.0 mL, respectively. After standing for 1 h, 4MBA molecules were linked with the Au@Ag core–shell NPs to form the 4MBA-labelled Au@Ag core–shell NPs (Au@Ag@4MBA NPs). Then, the prepared Au@Ag@4MBA NPs solutions were centrifuged at 8000 rpm for 15 min and the sediments were dispersed in 2.0 mL deionized water under ultrasonic oscillation, respectively. Next, 1.0 μL of EDC solution (10 mM, PBS solvent) was added to 1.0 μL of NHS solution (10 mM, dissolved in PBS) to prepare an EDC/NHS mixture solution (2.0 μL, 10 mM). Then, at room temperature, the EDC/NHS mixture solution was added to 2.0 mL of the Au@Ag@4MBA NPs solution and reacted for 40 min to obtain the carboxyl-activated Au@Ag@4MBA NPs. Then, 500 μL of the carboxyl-activated Au@Ag@4MBA NPs solution was mixed with 50 μL of 5′-NH2-ssDNA solution (10 μM, TE solvent) and incubated at 25 °C for 5 h to form the Au@Ag@4MBA@5′-NH2-ssDNA probes. Subsequently, the probes solution was purified by of centrifugation at 2500 rpm three times for 15 min each time. Finally, the sediment was dispersed in 500 μL of PBS solution under ultrasonic oscillation to obtain the subsequent bioassay.
2.3 Preparation of the Ag/TiO2@3′-NH2-ssDNA substrate
The Ag/TiO2@3′-NH2-ssDNA substrate was prepared by the following steps. First, a 0.20 mm thickness Ti sheet with an area of 2 cm2 was put into deionized water for ultrasonic washing for 10 min to remove surface impurities. Next, the Ti sheet was fast immersed in a diluted hydrofluoric acid solution (HF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O = 1:30) to remove surface oxides during the process of surface acidification, and then instantly ultrasonically cleaned in an aqueous solution for 30 min. Then, NaOH solution (20 mL, 2.50 M) was poured into a 25 mL of polytetrafluoroethylene hydrothermal autoclave liner, and the clean Ti sheet was immersed in the NaOH solution at a certain inclination angle for ensuring the full chemical reaction of both sides of the sheet, and then the sealed hydrothermal autoclave was placed into a vacuum drying oven at a 150 °C hot environment for 16 h. Subsequently, the hydrothermally treated Ti sheet was put into a clean polytetrafluoroethylene hydrothermal autoclave liner containing deionized water at 150 °C for 5 h. Thus far, the TiO2 NWs structures were formed on the surface of the Ti sheet. Second, Ag was deposited on the TiO2 NWs to prepare the Ag/TiO2 NWs substrate by magnetron sputtering under optimal conditions (5 × 10−4 Pa background vacuum, 0.5 Pa sputtering pressure, 20 sccm argon flow and 30 W radio frequency target sputtering power). Third, 3′-NH2-ssDNA was immobilized on the surface of the Ag/TiO2 NWs substrate by three chemical processes: (1) carboxylation of the substrate: the Ag/TiO2 NWs substrate was immersed into 2.0 mL of the mixed solution of 100 mM MUA and 100 mM MU and left overnight; (2) activation of the carboxyl groups: the carboxylated Ag/TiO2 NWs substrate was immersed into the mixed solution of 50 mM NHS and 200 mM EDC and leftovernight; (3) linking of DNA: the carboxyl-activated Ag/TiO2 NWs substrate was immersed into the PBS solution containing 1.0 μM of the 3′-NH2-ssDNA and left overnight. Then, the as-prepared Ag/TiO2@3′-NH2-ssDNA substrate was successively washed using PBS and deionized water for three cycle times. Finally, the Ag/TiO2@3′-NH2-ssDNA substrate was successfully obtained and stored in PBS solution for later SERS detection.
:H2O = 1:30) to remove surface oxides during the process of surface acidification, and then instantly ultrasonically cleaned in an aqueous solution for 30 min. Then, NaOH solution (20 mL, 2.50 M) was poured into a 25 mL of polytetrafluoroethylene hydrothermal autoclave liner, and the clean Ti sheet was immersed in the NaOH solution at a certain inclination angle for ensuring the full chemical reaction of both sides of the sheet, and then the sealed hydrothermal autoclave was placed into a vacuum drying oven at a 150 °C hot environment for 16 h. Subsequently, the hydrothermally treated Ti sheet was put into a clean polytetrafluoroethylene hydrothermal autoclave liner containing deionized water at 150 °C for 5 h. Thus far, the TiO2 NWs structures were formed on the surface of the Ti sheet. Second, Ag was deposited on the TiO2 NWs to prepare the Ag/TiO2 NWs substrate by magnetron sputtering under optimal conditions (5 × 10−4 Pa background vacuum, 0.5 Pa sputtering pressure, 20 sccm argon flow and 30 W radio frequency target sputtering power). Third, 3′-NH2-ssDNA was immobilized on the surface of the Ag/TiO2 NWs substrate by three chemical processes: (1) carboxylation of the substrate: the Ag/TiO2 NWs substrate was immersed into 2.0 mL of the mixed solution of 100 mM MUA and 100 mM MU and left overnight; (2) activation of the carboxyl groups: the carboxylated Ag/TiO2 NWs substrate was immersed into the mixed solution of 50 mM NHS and 200 mM EDC and leftovernight; (3) linking of DNA: the carboxyl-activated Ag/TiO2 NWs substrate was immersed into the PBS solution containing 1.0 μM of the 3′-NH2-ssDNA and left overnight. Then, the as-prepared Ag/TiO2@3′-NH2-ssDNA substrate was successively washed using PBS and deionized water for three cycle times. Finally, the Ag/TiO2@3′-NH2-ssDNA substrate was successfully obtained and stored in PBS solution for later SERS detection.
2.4 SERS-based bioassay protocol
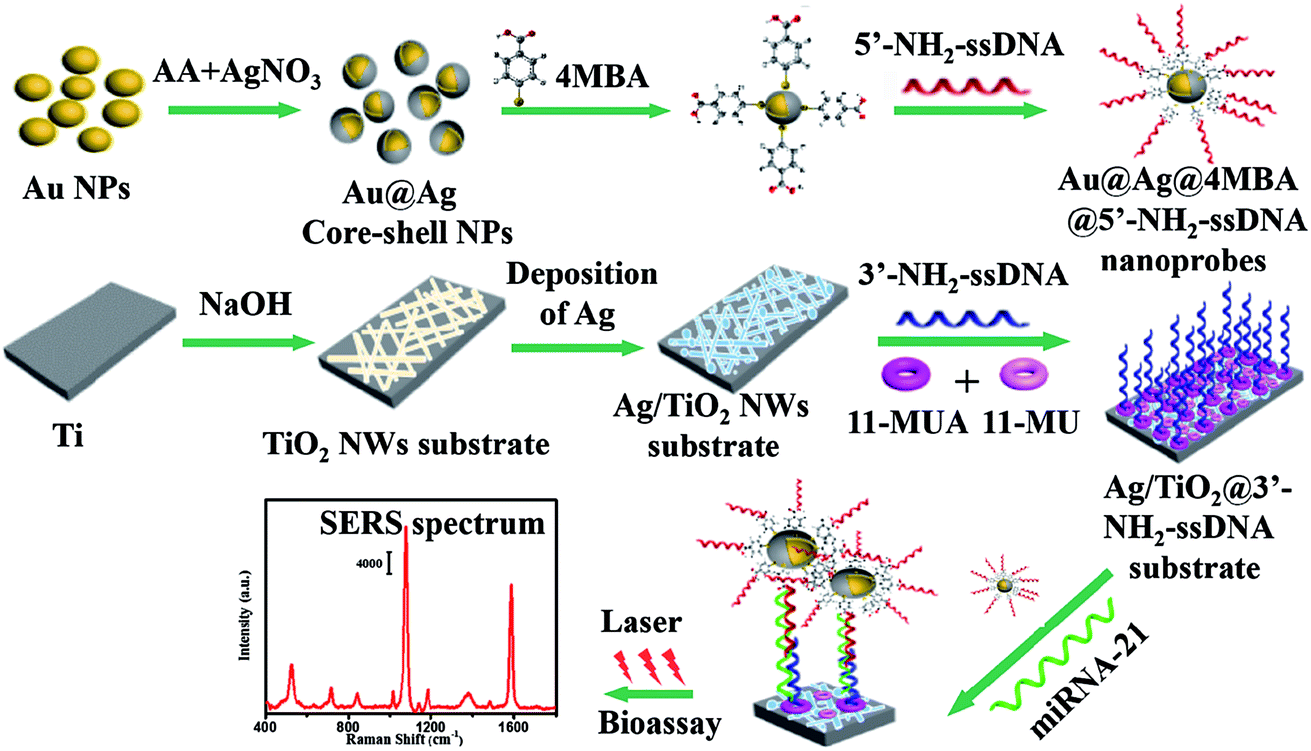
The SERS-based sandwich bioassay protocol is shown in Fig. 1. In our experiments, using DEPC-H2O as a buffer solution, the purchased miRNA-21 (1 OD) was diluted into seven target samples from 1.0 fM to 1.0 nM with a concentration gradient of ten times. Taking a target sample as an example, 30 μL of miRNA-21 and 50 μL of Au@Ag@4MBA@5′-NH2-ssDNA probes were mixed together and incubated at 37 °C for 1 h. Next, under shaking of 250 rpm, the as-fabricated Ag/TiO2@3′-NH2-ssDNA substrate was immersed in the mixed solutions at 37 °C for 2 h. Then, the sandwich complex composed of the Au@Ag@4MBA@5′-NH2-ssDNA probes, miRNA-21 and the Ag/TiO2@3′-NH2-ssDNA substrate was formed by the complementary base pairing of DNA. Finally, after washing three cycle times in turn by PBS and deionized water, the sandwich complex was used for the SERS measurement.
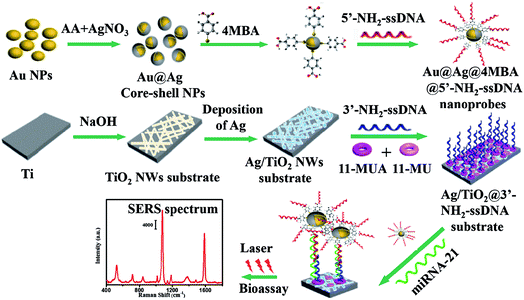 |
| | Fig. 1 Schematic of the SERS-based sandwich bioassay protocol. | |
2.5 Instruments
Scanning electron microscopy (SEM) images were obtained on a field-emission scanning electron microscope (SU-70, Hitachi, Japan) operating at 10 kV accelerating voltage. Transmission electron microscopy (TEM) images were obtained using a TEM (JEM-2100F, JEOL, Japan) operating at an accelerating voltage of 200 kV. Ag was deposited by the magnetron sputtering equipment (TA13-XD, Kurt J., Lesker, USA). The X-ray diffraction (XRD) patterns were measured using an X-ray diffractometer (D8 Discover, Bruker, Germany). Optical absorption spectra were measured with a UV-vis spectrometer (TU1901, Pgeneral, China). ζ-Potential measurements were performed with a nanoparticle size analyzer (Zetasizer Nano S90, Malvern, UK). A high-speed centrifuge (TGL-20M, Lu Xiangyi Centrifuge Instrument Co. Ltd, China) was used for sample purification. A constant temperature mixer (T-MSC, Topscien, Canada) was used for DNA hybridization. An RT-qPCR system (StepOne Plus, ABI, USA) was used for the quantitative detection of microRNA. An injection pump (AL-4000, World Precision Instruments Inc, China) was used to control the dropping speed of the reaction solution. SERS signals were measured using a miniature Raman spectrometer (BWS415, B&W Tek Inc., USA), which was equipped with a semiconductor laser (785 nm, 499.95 mW), a dispersed grating of 1200 lines mm−1 and a charge-coupled device (CCD) (2048 × 2048 pixels) detector. All the analyses were performed at room temperature.
3. Results and discussion
3.1 Characterization of the probes
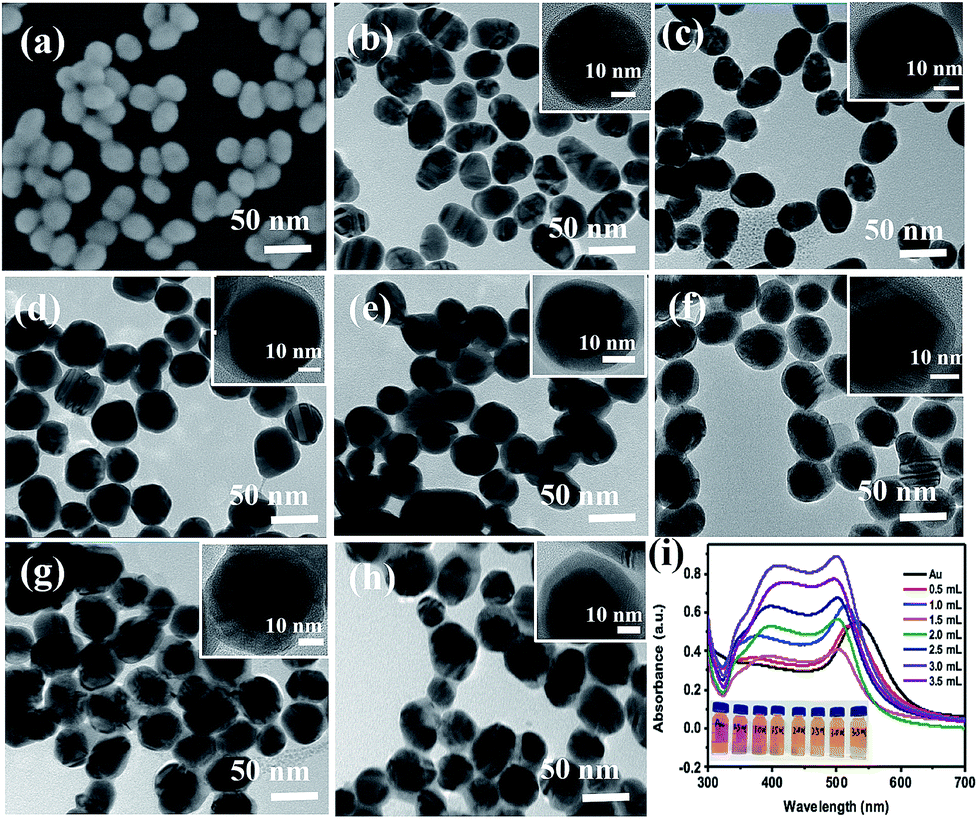
The SEM image of the prepared Au NPs and the TEM images of the synthesized Au@Ag core–shell NPs are shown in Fig. 2. It could be easily found from Fig. 2(a) that the Au NPs display an approximate spherical morphology and a uniform size of 35 ± 5 nm. Also, Fig. 2(b–h) clearly exhibit that the Au@Ag core–shell NPs have a spherical core–shell structure and good monodispersity and uniformity. The insets in Fig. 2 show that the thicknesses of Ag shells gradually increase from 1.0 nm to 10.0 nm with increasing the AgNO3 amount from 0.50 to 3.50 mL. As shown in Fig. 2(i), two absorption peaks appear in the absorption spectra also demonstrate the structures of Au core and Ag shell for the synthesized nanoparticles. Also, with increasing the Ag shell thickness, the absorption peaks of the Au core and Ag shell respectively exhibit a blue-shift from 534 to 498 nm and a red-shift from 370 to 410 nm due to the plasmonic coupling effect between the Au core and Ag shell.35,36In fact, as the inset shows, with increasing the Ag mole fraction, the colours of the Au@Ag core–shell NPs solutions show a distinguishable change from wine red to orange, which is consistent with the absorption spectra of the nanoparticle solutions and could be attributed to the composition, size and morphology-dependent surface plasmon resonance.37 Clearly, the optical property of the Au@Ag core–shell NPs could be easily tuned over a wide visible spectral range by simply changing the Ag shell thickness.
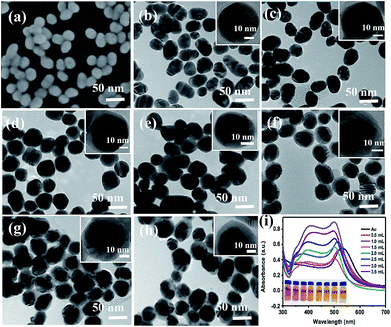 |
| | Fig. 2 (a) SEM image of Au NPs and (b–h) TEM images of the Au@Ag core–shell NPs synthesized under adding AgNO3 in amounts from 0.50 to 3.50 mL at a gradient of 0.50 mL, respectively. The inset HRTEM images present the thickness of Ag shell from 1.0 to 10.0 nm; (i) the absorption spectra of Au NPs and Au@Ag core–shell NPs with the increase in the amount of AgNO3 from 0.50 to 3.50 mL, the inset shows the colour of the solutions at different dosages of AgNO3 solution. | |
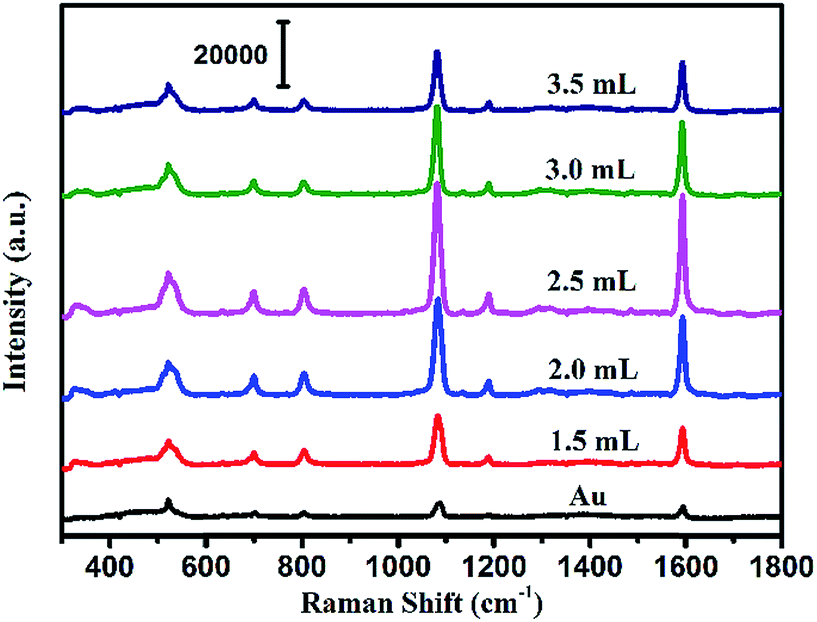
To evaluate the SERS performances of the Au@Ag core–shell NPs, the SERS spectra of Au@Ag@4MBA NPs were measured and are shown in Fig. 3. It could be found that the SERS spectra of 4MBA showed evident fingerprint characteristics. One of the predominant bands was located at 1078 cm−1, which was ascribed to the aromatic ring breathing mode coupled with symmetric γ(C–H) and ν(C–S); while another band located at 1588 cm−1 was assigned to the mode of asymmetric γ(C–H) and ν (CC).38 As shown in Fig. 3, the intensities of SERS peaks increase continuously with increasing the AgNO3 amount to 2.50 mL up to a maximum and then decrease. In fact, the excellent SERS enhancement properties of the Au@Ag core–shell NPs originate from the synergistic plasmonic enhancement of the Au core and the Ag shell, and the plasmon resonance coupling effect between the Au cores and Ag shell is enhanced with the increases in the Ag shell thickness. However, for a thick Ag shell, the plasmon resonance of the gold core is covered and the plasmon resonance of Ag shell dominates, which causes the SERS effect to become weak.39 In our experiment, when the amount of AgNO3 was 2.50 mL, that is, the Ag shell thickness was about 7.0 nm, the Au@Ag core–shell NPs showed the best SERS enhancement effect. Thus, the Au@Ag core–shell NPs with 7.0 nm Ag shell thickness were selected as an optimal precursor and used in all the later experiments.
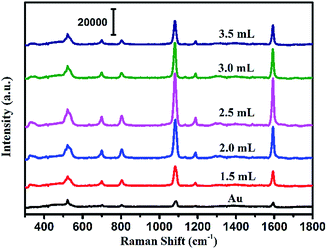 |
| | Fig. 3 SERS spectra of the Au NPs and Au@Ag core–shell NPs with the increase in the amount of AgNO3 added to the solution. | |
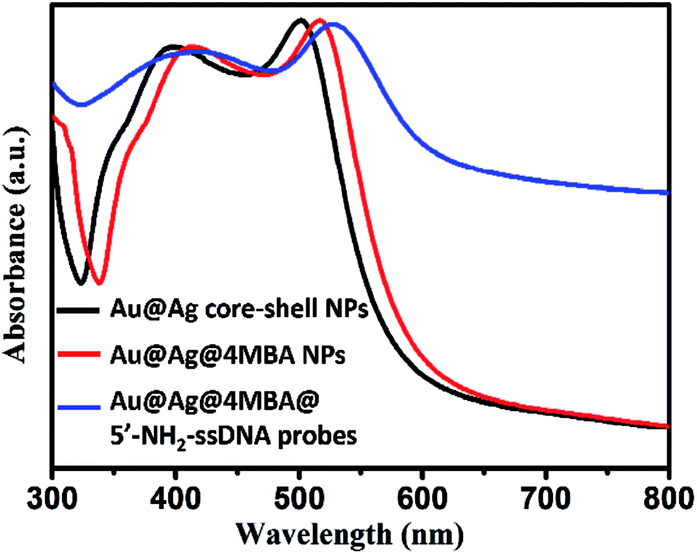
Furthermore, the formation processes of the Au@Ag@4MBA@5′-NH2-ssDNA probes were investigated by UV-vis absorption spectroscopy and ζ-potential measurements. As shown in Fig. 4, compared to the absorption spectrum of the Au@Ag core–shell NPs, the absorption band of Au@Ag@4MBA NPs was broadened and red-shifted, which indicate the successful coating of 4MBA on the surfaces of the Au@Ag core–shell NPs.40 Also, the absorption band of the Au@Ag@4MBA@5′-NH2-ssDNA probes was further broadened and red-shifted because the size and the aggregation degree of the probes increased after adding 5′-NH2-ssDNA.41 Meanwhile, the surface-modification properties of the Au@Ag core–shell NPs were analyzed by ζ-potential measurements. As listed in Table 2, during the immobilization of 4MBA molecules, the ζ-potential of the Au@Ag core–shell NPs changed from −19.3 to −25.9 mV due to the formation of the negatively charged carboxyl group of 4MBA around the Ag shell. After the 5′-NH2-ssDNA were linked on the surface of Au@Ag@4MBA NPs, the surface potential of the Au@Ag@4MBA@5′-NH2-ssDNA probes was −17.1 mV owing to the positively charged amino group of the 5′-NH2-ssDNA.42 This displays that 4MBA and 5′-NH2-ssDNA were successively immobilized on the surfaces of Au@Ag core–shell NPs.
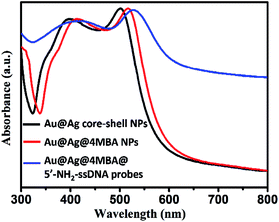 |
| | Fig. 4 UV-vis absorption spectra of the Au@Ag core–shell NPs, Au@Ag@4MBA NPs and Au@Ag@4MBA@5′-NH2-ssDNA probes. | |
Table 2
ζ-Potential measurements
| Sample |
Zeta-potential (mV) |
| Au@Ag core–shell NPs |
−19.3 ± 0.32 |
| Au@Ag@4MBA NPs |
−25.9 ± 0.53 |
| Au@Ag@4MBA@5′-NH2-ssDNA probes |
−17.1 ± 0.23 |
3.2. Characterization of the Ag/TiO2 NWs substrates
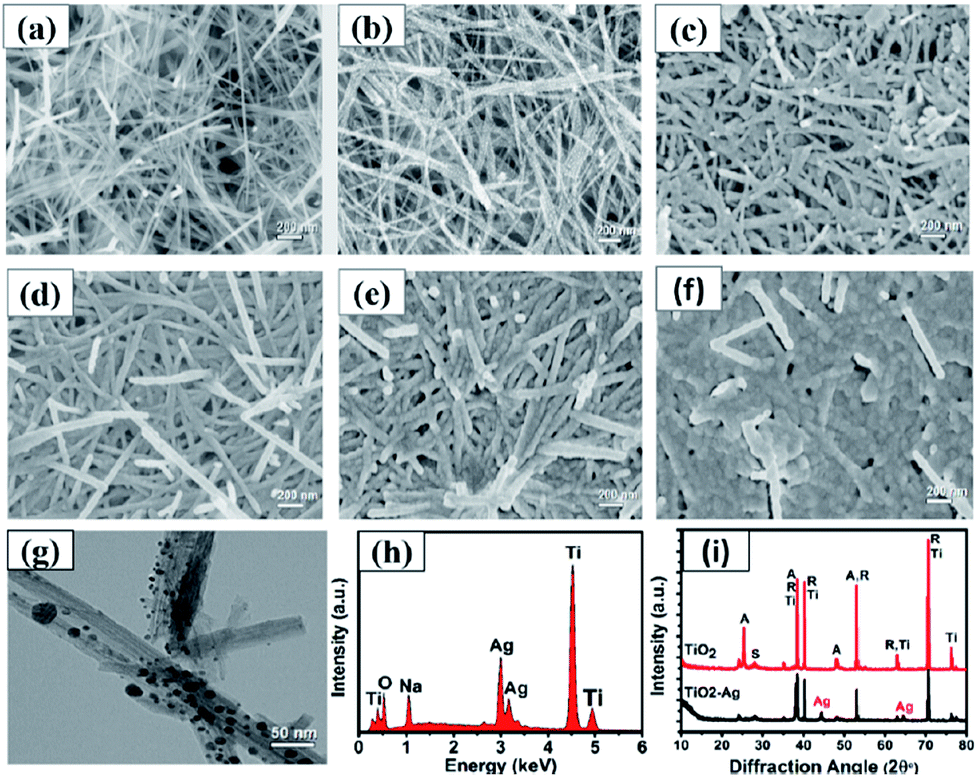
As shown in Fig. 5, the morphology and composition of the fabricated TiO2 NWs and Ag/TiO2 NWs substrates were analyzed by SEM, TEM, EDS and XRD. It could be observed from Fig. 5(a) that the TiO2 NWs have wire-like morphologies with a micron-level length and a thin cross-section. Fig. 5(b–f) show that the Ag NPs were uniformly decorated on the surfaces of TiO2 NWs and the density of Ag NPs became larger and larger up to them forming a thin silver layer with increasing the sputtering time. Fig. 5(g) clearly shows that the average size of the Ag NPs deposited on the surface of TiO2 NWs was nearly 10 nm under our sputtering deposition conditions. Moreover, the EDXS spectrum (Fig. 5(h)) illustrates that Ag, Ti, O and Na elements existed in the Ag/TiO2 NWs substrate and the elemental compositions (atomic percentage) constituted by Ti, O and Ag were 44.11, 36.88 and 9.35, respectively. Here, the Na element comes from the residual NaTiO3 obtained in the hydrothermal treatment process of Ti sheet and NaOH. In addition, the XRD patterns (Fig. 5(i)) display that the TiO2 NWs have two phase structures, i.e. anatase and rutile, while Ag is a single crystal.
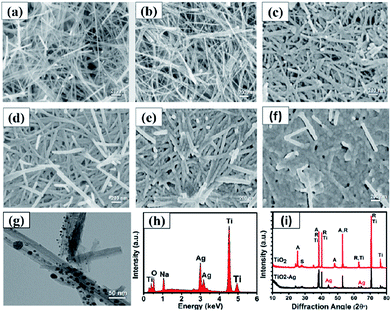 |
| | Fig. 5 SEM images of (a) TiO2 NWs and Ag/TiO2 NWs substrates prepared at different sputtering times: (b) 1, (c) 2, (d) 3, (e) 4, (f) 5 min; (g) TEM image of Ag/TiO2 NWs; (h) EDXS spectrum of Ag/TiO2 NWs substrate; (i) XRD spectra of TiO2 NWs and Ag/TiO2 NWs substrate (A, R, S, Ti and Ag represent anatase, rutile, sodium titanate, titanium and silver, respectively). | |
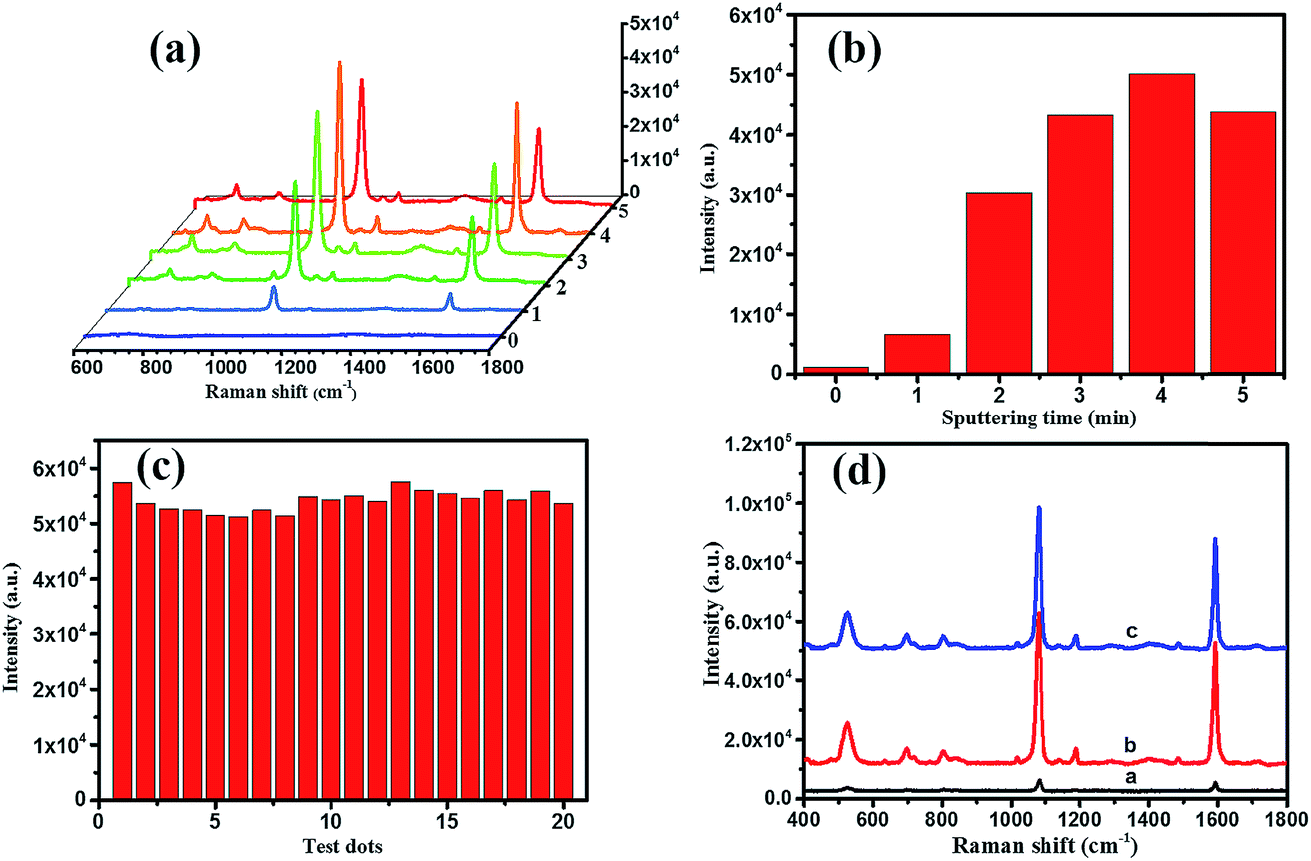
On the other hand, for evaluating the SERS performances of the Ag/TiO2 NWs substrates, the SERS spectra of 4MBA adsorbed on the surface of substrates were measured with a 785 nm laser excitation. Fig. 6(a) shows that the SERS performances of the Ag/TiO2 NWs substrates were significantly better than those of the TiO2 NWs substrates due to the electromagnetic enhanced effect of the Ag NPs, besides the chemical enhancement derived from the charge-transfer effect among Ag NPs, TiO2 NWs and 4MBA molecules.43 Also, Fig. 6(b) visually displays that the SERS peak intensities at 1078 cm−1 first increase with increasing the sputtering time from 0 to 4 min and then decrease at 5 min. This illustrates that the Ag NPs get closer together so that there are more “hot-spots” produced to contribute to the enhanced electromagnetic effect.44 However, with an overlong sputtering time, the Ag NPs join together and even form a film, leading to a drop in SERS performance due to the decrease in the number of “hot-spots”. Thus, the Ag/TiO2 NWs substrate prepared at a sputtering time of 4 min exhibited the highest SERS enhancement ability and was thus selected as the optimal SERS-active substrate to be used in the later experiments.
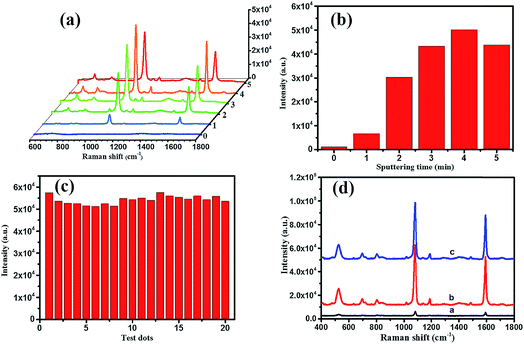 |
| | Fig. 6 (a) SERS spectra and (b) the intensities of SERS peaks at 1078 cm−1 of 4MBA (10 mM) dropped on the surfaces of Ag/TiO2 NWs substrates prepared at different sputtering times; (c) the intensities of the SERS peak at 1078 cm−1 collected from 20 random testing spots on the optimal SERS-active substrate; (d) Raman spectra of 4MBA powder on a quartz slide (line a), the freshly prepared substrate (line b) and the saved substrate (line c). | |
In addition, as shown in Fig. 6(c), the intensities of the SERS peak at 1078 cm−1 of 4MBA located at 20 random testing spots on the surface of optimal SERS-active substrate exhibited a small fluctuation, which showed the good uniformity of the Ag/TiO2 NWs SERS-active substrate. After saving for two months, the SERS spectrum of the substrate (line c in Fig. 6(d)) showed a slight decline compared with that of the freshly prepared substrate (line b), which illustrated the good stability of the Ag/TiO2 NWs SERS-active substrate. Furthermore, the Raman enhancement factor (EF) of the optimal SERS-active substrate was determined using the following equation:45–47
| EF = (ISERS/Ibulk) × (Nbulk/NSERS) |
where
ISERS and
Ibulk are the integral intensity of the SERS peak of 4MBA molecules adsorbed on the Ag/TiO
2 NWs substrate and the Raman spectrum of 4MBA powder and
NSERS and
Nbulk are the number of 4MBA molecules within the laser spot in the presence and absence of the Ag/TiO
2 NWs substrate, respectively. According to the SERS spectra (line b) in
Fig. 6(d), the value of
ISERS and
Ibulk corresponding to the SERS peak at 1078 cm
−1 were calculated to be 8.87 × 10
5 and 2.35 × 10
4, respectively. Taking in to consideration the specifications of the Raman spectrometer, the diameter of the laser spot was calculated to be 2.39 μm by using the equation:
46Ddiameter = (
λ/NA) × 1.22, in which
λ and NA are 785 nm and 0.4, respectively.
NSERS was determined to be 1.35 × 10
7 by using the equation
NSERS =
NA ×
A/
δ, where
NA is the Avogadro constant, the effective area occupied by 4MBA molecules within the laser spot
A = 4.499 μm
2 and the area of 4MBA molecules per mole in a self-assembled monolayer is
δ = 2.0 × 10
9 cm
2 mol
−1. The equation
Ddepth = 2
π/(NA)
2 was used to calculate the penetration depth of the focused laser spot.
47 Then, the irradiated volume of 4MBA was 14.7 μm
3 and
Nbulk = 8.85 × 10
11 was calculated by using a powder density of 1.5 g cm
−3 and molecular weight of 154.19 g mol
−1. Finally, the EF of the Ag/TiO
2 NWs SERS-active substrate was obtained as 2.05 × 10
6. This reveals that the fabricated Ag/TiO
2 NWs substrate demonstrated remarkable SERS activity, making them suitable for effective Raman detection.
3.3 SERS-based bioassay of miRNA-21
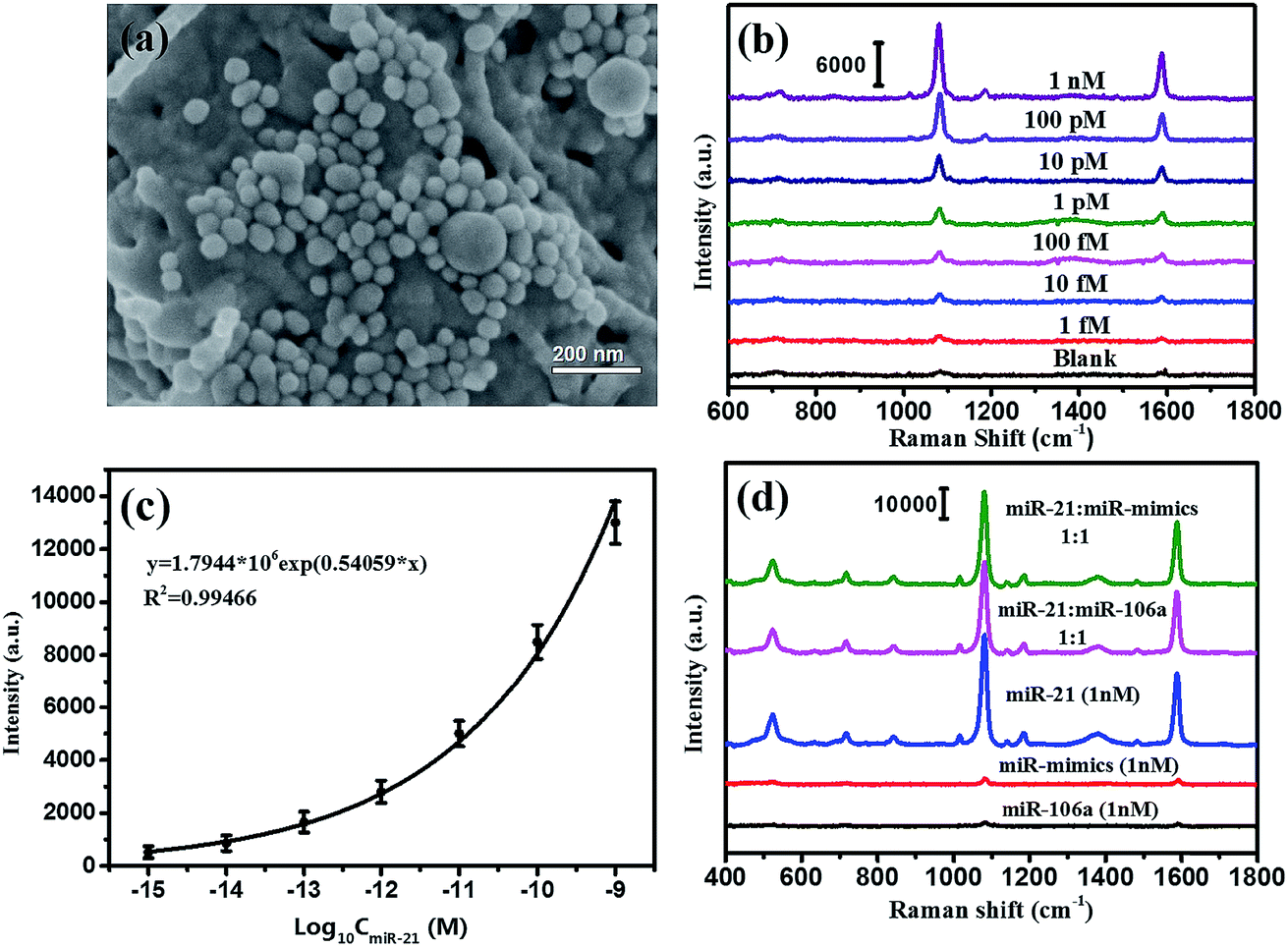
According to the SERS-based bioassay protocol described in Section 2.4, miRNA-21 was detected by measuring the SERS signal of 4MBA in the sandwich structure under laser irradiation with 785 nm wavelength. As shown in Fig. 7(a), the Au@Ag@4MBA@5′-NH2-ssDNA probes were distributed on the Ag/TiO2 NWs@3′-NH2-ssDNA substrate dispersedly, and the sandwich complex as the bioassay platform was formed in the presence of the target nucleotide miRNA-21. Fig. 7(b) shows the SERS spectra corresponding to miRNA-21 in the range of concentrations from 1.0 fM to 1.0 nM with a gradient of 10 times. This indicates that the intensities of the SERS peaks decline with decreasing the miRNA-21 concentration, and that the weakest SERS peaks corresponding to the 1.0 fM miRNA-21 could still be distinguished from the SERS peaks of the blank control sample without miRNA-21. Fig. 7(c) shows that the intensities of SERS peaks at 1078 cm−1 were dependent on the concentration of miRNA-21, and the fitting curve satisfied the equation y = 1.7944 × 106exp(0.54059x) with a correlation coefficient of R2 = 0.99466. From the standard dose–response curve, the limit of detection (LOD) of miRNA-21 was estimated to be 0.75 fM by considering the blank determination method.48 The analytical performances of the different assays are compared in Table 3. Furthermore, the specificity of the bioassay was investigated by replacing miRNA-21 with an equivalent amount of noncomplementary miRNAs (miR-106a and miR-mimics) following the same bioassay protocol. As shown in Fig. 7(d), strong SERS signals were only generated in the bioassay of the miRNA-21 samples, but no SERS peak was observed from the SERS spectra corresponding to the noncomplementary miRNAs (black curve and red curve). Thus, the constructed SERS-based sandwich bioassay platform exhibited a high specificity for detecting miRNA-21.
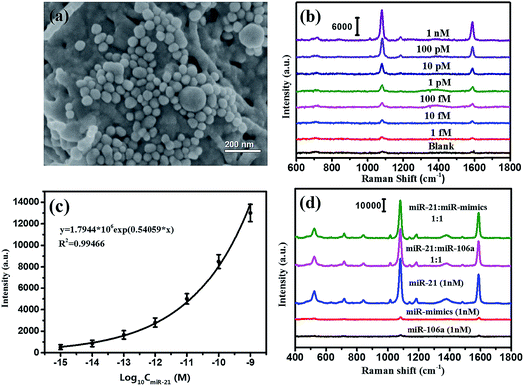 |
| | Fig. 7 (a) SEM images of the sandwich bioassay complex, (b) SERS spectra of 4MBA corresponding to the different concentrations of miRNA-21, (c) the standard dose–response curves of miRNA-21 with logarithmic concentrations, (d) SERS spectra of the sandwich bioassay for miRNA-21, miR-106a, miR-mimics at the same concentration of 1 nM and their mixture solutions with a concentration ratio of 1:1. | |
Table 3 Comparison between the proposed assay and other reported methods for miRNA detection
| Methods |
Linear range (pM) |
LOD (fM) |
References |
| Fluorescence method based on gold-nanorod-functionalized polydiacetylene microtube waveguide |
50–1000 |
10 |
(Zhu, 2016)49 |
| Electrochemical sensor based on tungsten oxide-graphene composites coupled with a catalyzed hairpin assembly |
0.0001–100 |
0.05 |
(Huang, 2016)18 |
| A new enzyme-free quadratic SERS signal amplification approach |
0.001–10 |
0.3 |
(Zheng, 2015)50 |
| Au@Ag core–shell NPs and Ag/TiO2 NWs substrate |
0.001–100000 |
0.75 |
This work |
Finally, the reliability of the SERS-based sandwich bioassay protocol was checked by the test data using RT-qPCR technology. As listed in Table 4, the test results of the SERS-based bioassay protocol were not only consistent with those of RT-qPCR, but it also displayed higher detection sensitivities. Therefore, the proposed SERS-based sandwich bioassay protocol is a valid strategy for detecting miRNA-21 and has good application potential for cancer diagnoses.
Table 4 Detection results of the samples
| Sample |
SERS (fmol L−1) |
RT-qPCR (fmol L−1) |
Relative deviation |
| 1 |
1750.000 |
1661.448 |
5.32% |
| 2 |
395.639 |
422.262 |
−6.30% |
| 3 |
201.202 |
237.114 |
−15.1% |
| 4 |
83.365 |
— |
— |
| 5 |
12.564 |
— |
— |
| 6 |
2.064 |
— |
— |
4. Conclusions
In this work, a novel SERS-based sandwich bioassay protocol was developed and applied to detect the concentrations of miRNA-21 by a sandwich complex composed of Au@Ag@4MBA@5′-NH2-ssDNA probes and a Ag/TiO2@3′-NH2-ssDNA substrate. By measuring the SERS signals of 4MBA in the probes, the concentrations of miRNA-21 in the samples were quantitatively analyzed. The experimental results showed the proposed bioassay strategy displayed a high detection sensitivity for miRNA-21 in a wide detection range. Also, control experiments exhibited the superior biological specificity of the SERS-based sandwich bioassay platform for detecting miRNA-21. In addition, the reliability of the proposed SERS-based sandwich bioassay protocol was verified by the RT-qPCR method via comparing their test data of miRNA-21. Further, by adjusting the sequences of the complementary DNA single chain decorated on the probes and the substrate, our detection protocol may be extended to the detection of various miRNA. It is expected that the SERS-based sandwich bioassay protocol has favourable application prospects in the early diagnosis of cancer.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 61320106014, 61675104 and 61704095). This work was also partially supported by the Natural Science Foundation of Zhejiang Province (Grant no. LY19F050002), the Natural Science Foundation of Ningbo (Grant No. 2018A610316) and K. C. Wong Magna Fund in Ningbo University, China.
References
- J. Ferlay, H. R. Shin, F. Bray, D. Forman, C. Mathers and D. M. Parkin, Int. J. Cancer, 2010, 127, 2893–2917 CrossRef CAS PubMed.
-
B. W. Stewart and C. P. Wild, World Cancer Report 2014, IARC Nonserial Publication, Lyon, 2014 Search PubMed.
- G. Bastarrika, M. J. Garcia-Velloso, M. D. Lozano, U. Montes, W. Torre, N. Spiteri, A. Campo, L. Seijo, A. B. Alcaide, J. Pueyo, D. Cano, I. Vivas, O. Cosin, P. Dominguez, P. Serra, J. A. Richter, L. Montuenga and J. J. Zulueta, Am. J. Respir. Crit. Care Med., 2005, 171, 1378–1383 CrossRef PubMed.
- C. Liu, X. Yan, X. Zhang, W. Yang, W. Peng, D. Shi, P. Zhu, W. Huang and Q. Yuan, Phys. Med. Biol., 2007, 52, 419–427 CrossRef PubMed.
- H. Saisho and T. Yamaguchi, Pancreas, 2004, 28, 273–278 CrossRef PubMed.
- C. Liu, Y. Yang and Y. Wu, AAPS J., 2018, 20, 41 CrossRef PubMed.
- R. J. Henderson, J. A. Eastham, J. D. Culkin, T. Whatley, J. Mata, D. Venable and O. Sartor, JNCI, J. Natl. Cancer Inst., 1997, 89, 134–138 CrossRef CAS PubMed.
- A. Cedrone, M. Covino, E. Caturelli, M. Pompili, G. Lorenelli, M. R. Villani, D. Valle, M. Sperandeo, G. L. Rapaccini and G. Gasbarrini, Hepato-Gastroenterology, 2000, 47, 1654–1658 CAS.
- M. Yasue, J. Sakamoto, S. Teramukai, T. Morimoto, K. Yasui, N. Kuno and Y. Ohashi, Pancreas, 1994, 9, 735–740 CrossRef CAS PubMed.
- I. Raiko, I. Sander, D. G. Weber, M. Raulf-Heimsoth, A. Gillissen, J. Kollmeier and G. Johnen, BMC Cancer, 2010, 10, 242 CrossRef PubMed.
- V. Dutoit-Lefèvre, S. Dubucquoi, D. Launay, V. Sobanski, P. Dussart, P. Chafey and D. Lefranc, PLoS One, 2015, 10, e0132142 CrossRef PubMed.
- X. G. Li, M. Meng, L. Zheng, Z. Xu, P. Song and R. Xi, Anal. Chem., 2016, 88, 8556–8561 CrossRef CAS PubMed.
- T. A. Farazi, J. I. Spitzer, P. Morozov and T. J. Tuschl, J. Pathol., 2011, 223, 102–115 CrossRef CAS PubMed.
- C. L. Yauk, A. Rowan-Carroll, J. D. Stead and A. Williams, BMC Genomics, 2010, 11, 330 CrossRef PubMed.
- Y. X. Chen, K. J. Huang and K. X. Niu, Biosens. Bioelectron., 2018, 99, 612–624 CrossRef CAS PubMed.
- M. Z. Micheal, S. M. O'Connor, N. G. Van holst pellekan, G. P. Young and R. J. James, Mol. Cancer Res., 2003, 1, 882–891 Search PubMed.
- D. Wen, D. Pan, P. Lin, Q. Mo, Y. Wei, Y. Luo and H. Yang, Mol. Med. Rep., 2018, 18, 2631–2642 CAS.
- H. L. Shuai, K. J. Huang, L. L. Xing and Y. X. Chen, Biosens. Bioelectron., 2016, 86, 337–345 CrossRef CAS PubMed.
- Y. X. Chen, W. J. Zhang, K. J. Huang, M. b. Zheng and Y. C. Mao, Analyst, 2017, 142, 4843 RSC.
- A. Markou, E. G. Tsaroucha and L. Kaklamanis, Clin. Chem., 2008, 54, 1696–1704 CrossRef CAS PubMed.
- G. Song, H. Zhou, J. Gu, Q. Liu, W. Zhang, H. Su and D. Zhang, J. Mater. Chem. B, 2017, 5, 1594–1600 RSC.
- C. W. Lee and F. G. Tseng, Biomicrofluidics, 2018, 12, 011502 CrossRef PubMed.
- L. Bi, Y. Wang, Y. Yang, Y. Li, S. Mo, Q. Zheng and L. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 15381–15387 CrossRef CAS PubMed.
- D. Chen, J. Zhou, M. Rippa and L. Petti, J. Appl. Phys., 2015, 118, 163101 CrossRef.
- D. Y. Wu, X. M. Liu, S. Duan, X. Xu, B. Ren, S. H. Lin and Z. Q. Tian, J. Phys. Chem. C, 2008, 112, 4195–4204 CrossRef CAS.
- T. Y. Olson, A. M. Schwartzberg, C. A. Orme, C. E. Talley, B. O'Connell and J. Z. Zhang, J. Phys. Chem. C, 2008, 112, 6319–6329 CrossRef CAS.
- D. Ma, C. Huang, J. Zheng, J. Tang, J. Li, J. Yang and R. Yang, Biosens. Bioelectron., 2018, 101, 167–173 CrossRef CAS PubMed.
- H. Zhang, C. Fu, Y. Yi, X. Zhou, C. Zhou, G. Ying and Y. Zhu, Anal. Methods, 2018, 10, 624–633 RSC.
- Y. Pang, C. Wang, J. Wang, Z. Sun, R. Xiao and S. Wang, Biosens. Bioelectron., 2016, 79, 574–580 CrossRef CAS PubMed.
- L. Cheng, R. A. Sharples and B. J. Scicluna, J. Extracell. Vesicles, 2014, 3, 17–27 Search PubMed.
- X. T. Wang, W. S. Shi, G. W. She and L. X. Mu, Phys. Chem. Chem. Phys., 2012, 14, 5891–5901 RSC.
- X. X. Zou, R. Silva, X. X. Huang, J. F. A. Sharab and T. Asefa, Chem. Commun., 2013, 49, 382–384 RSC.
- K. Y. B. Xie, Y. Y. Jin, Y. Z. Zhou and Y. Wang, Appl. Surf. Sci., 2014, 313, 549–557 CrossRef.
- B. H. Liu, G. M. Han, Z. P. Zhang, R. Y. Liu, C. L. Jiang, S. H. Wang and M. Y. Han, Anal. Chem., 2012, 84, 255–261 CrossRef CAS PubMed.
- Y. Sun, B. Wiley, Z. Y. Li and Y. J. Xia, J. Am. Chem. Soc., 2004, 126, 9399–9406 CrossRef CAS PubMed.
- S. Link, Z. L. Wang and M. A. El-Sayed, J. Phys. Chem. B, 1999, 103, 3529–3533 CrossRef CAS.
- Y. Q. Wang, B. Yan and L. X. Chen, Chem. Rev., 2013, 113, 1391–1428 CrossRef CAS PubMed.
- C. H. Ho and S. Lee, Colloids Surf., A, 2015, 474, 29–35 CrossRef CAS.
- O. Peña and U. Pal, Nanoscale Res. Lett., 2011, 6, 279–283 CrossRef PubMed.
- C. Song, Z. Wang, R. Zhang, J. Yang, X. Tan and Y. Cui, Biosens. Bioelectron., 2009, 25, 826–831 CrossRef CAS PubMed.
- C. Song, Z. Wang, J. Yang, R. Zhang and Y. Cui, Colloids Surf., B, 2010, 81, 285–288 CrossRef CAS PubMed.
- C. H. Wang, C. Yang, Y. Y. Song, W. Gao and X. Xia, Adv. Funct. Mater., 2005, 15, 1267–1275 CrossRef CAS.
- C. Yang, P. Liang, L. S. Tang and Y. F. Zhou, Appl. Surf. Sci., 2018, 436, 367–372 CrossRef CAS.
- A. Merlen, V. Gadenne, J. Ramann, V. Chevallier, L. Patrone and J. C. Valmalette, Nanotechnology, 2009, 20, 215705 CrossRef CAS PubMed.
- W. Li, P. H. C. Camargo, X. Lu and Y. Xia, Nano Lett., 2009, 118, 485–490 CrossRef PubMed.
- Y. He, S. Su, T. Xu, Y. Zhong, J. A. Zapien, J. Li, C. Fan and S. T. Lee, Nano Today, 2011, 2, 122–130 CrossRef.
- D. Chen, J. Zhou, M. Rippa and L. Petti, J. Appl. Phys., 2015, 118, 163101–163108 CrossRef.
- A. Shrivastava and V. Gupta, Chron. Young Sci., 2011, 2, 21–25 CrossRef.
- Y. Zhu, D. Qiu, G. Yang, M. Wang, Q. Zhang, P. Wang and J. R. Lakowicz, Biosens. Bioelectron., 2016, 85, 198–204 CrossRef CAS PubMed.
- J. Zheng, D. Ma, M. Shi, J. Bai, Y. Li, J. Yang and R. Yang, Chem. Commun., 2015, 51, 16271–16274 RSC.
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Zhaoheng
Liang
a,
Yaping
Zhang
b,
Lucia
Petti
*a,
Zhaoheng
Liang
a,
Yaping
Zhang
b,
Lucia
Petti