
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnalytical determination of heroin, fentanyl and fentalogues using high-performance liquid chromatography with diode array and amperometric detection†
Hadil M.
Elbardisy
 ab,
Christopher W.
Foster
a,
Loanda
Cumba
c,
Lysbeth H.
Antonides
a,
Nicolas
Gilbert
ad,
Christopher J.
Schofield
de,
Tarek S.
Belal
f,
Wael
Talaat
b,
Oliver B.
Sutcliffe
ad,
Hoda G.
Daabees
g and
Craig E.
Banks
*a
ab,
Christopher W.
Foster
a,
Loanda
Cumba
c,
Lysbeth H.
Antonides
a,
Nicolas
Gilbert
ad,
Christopher J.
Schofield
de,
Tarek S.
Belal
f,
Wael
Talaat
b,
Oliver B.
Sutcliffe
ad,
Hoda G.
Daabees
g and
Craig E.
Banks
*a
aFaculty of Science and Engineering, Manchester Metropolitan University, Chester Street, Manchester, M1 5GD, UK. E-mail: c.banks@mmu.ac.uk; Web: http://www.craigbanksresearch.com Fax: +44 (0)1612476831; Tel: +44 (0)1612471196
bPharmaceutical Analysis Department, Faculty of Pharmacy, Damanhour University, Damanhour, 22511, Egypt
cFaculty of Science & Health, Dublin City University, Glasnevin, Whitehall, Dublin 9, Ireland
dMANchester DRug Analysis and Knowledge Exchange (MANDRAKE), Faculty of Science and Engineering, Manchester Metropolitan University, Chester Street, Manchester, M1 5GD, UK
eGreater Manchester Police, Openshaw Complex, Lawton Street, Openshaw, Manchester, M11 2NS, UK
fDepartment of Pharmaceutical Analytical Chemistry, Faculty of Pharmacy, Alexandria University, Alexandria, 21521, Egypt
gPharmaceutical Chemistry Department, Faculty of Pharmacy, Damanhour University, Damanhour, 22511, Egypt
First published on 29th January 2019
Abstract
Over recent years there has been a progressive increase in the adulteration of common illicit street drugs, such as heroin and cocaine, with fentanyl and its derivatives (fentalogues) being the cause of over doses ending with fatal repercussions. Consequently, there is a need for the development of sensitive, selective and reliable analytical protocols for their separation and quantification. Herein, we report for the first time, a combination of high-performance liquid chromatography with a dual-diode array and electrochemical (amperometric) detector achieved for the simultaneous detection and quantification of heroin (HRN), fentanyl and ten fentalogues; the amperometric detection is achieved using a commercially available impinging jet flow-cell that incorporates in-house screen-printed graphite macroelectrodes (SPEs). Both protocols are analytically compared and contrasted in terms of their experimental parameters and chromatographic conditions with the separation and quantification being optimized, with these protocols demonstrating a high sensitivity and reproducibility. The proposed methods were successfully applied for the analysis of the investigated drugs of abuse, in the presence of common adulterants (e.g. caffeine, paracetamol and benzocaine), co-formulated excipients (starch, lactose, aerosil 200, etc.) and simultaneously within seized street samples.
Introduction
Fentanyl (Scheme 1), (N-phenyl-N-[1-(2-phenylethyl)piperidin-4-yl]propanamide, 2c) is a synthetic narcotic analgesic, a potent μ-opioid receptor agonist that is one hundred times more potent than heroin (HRN) (Scheme 1).1,2 Fentanyl was primarily introduced by Janssen Pharmaceutica in the early 1960's as an intravenous anesthetic during surgical operations.1 In the late 1980's, the same pharmaceutical company developed transdermal fentanyl patches under the trade name Duragesic® to relieve pain in cancer patients.3 It was not until the 1970's when the illicit use of “China Girl” or “China White” (alias given to fentanyl and its analogues) started to appear in California where it began to be substituted for HRN leading to unprecedented mortality rates.4 Over recent decades, adulteration of common illicit street drugs with fentanyl (owing to its low production cost, high potency and induced-euphoric properties) has made it a favorable “high” in the recreational drug market.5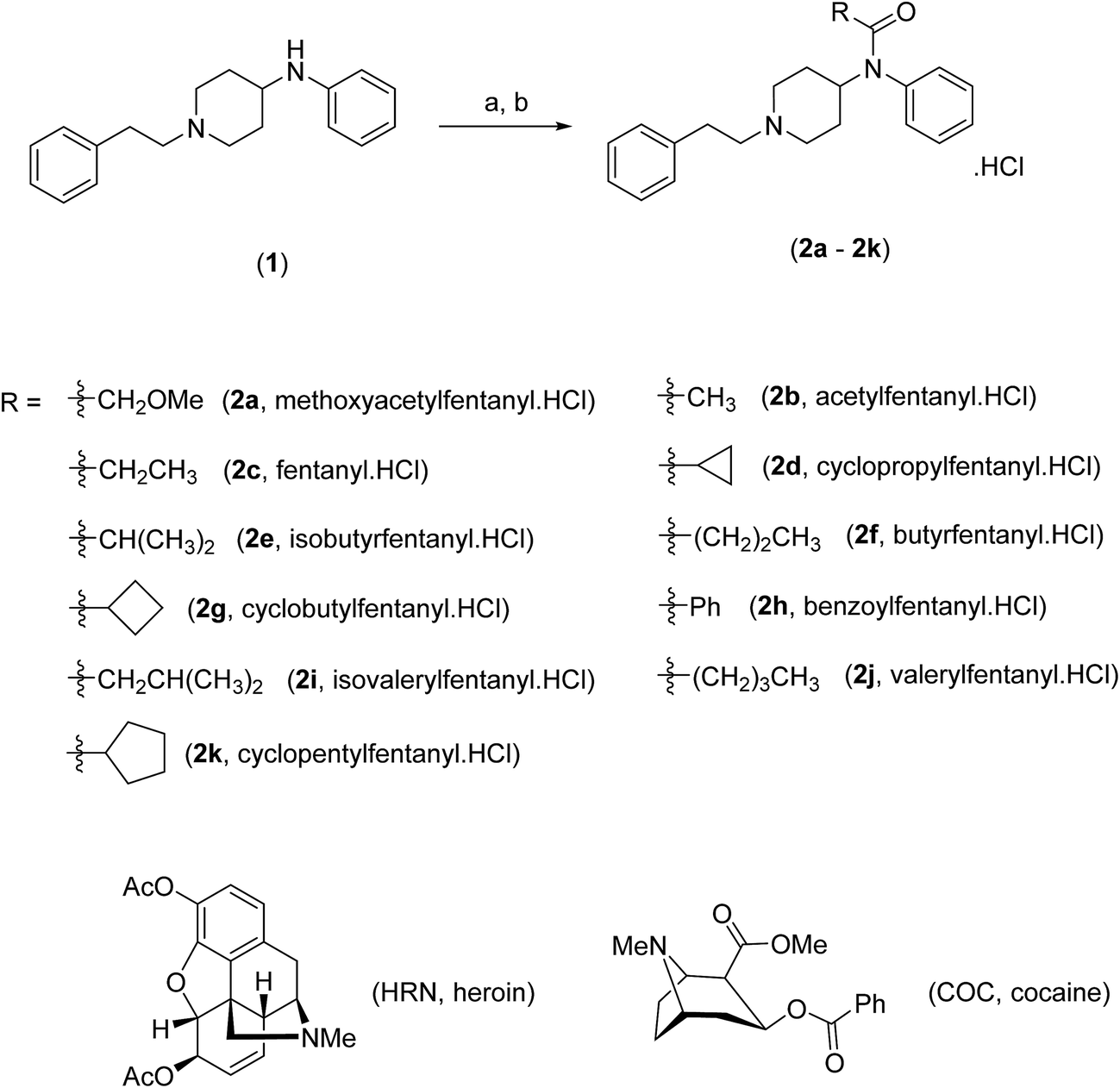 | ||
| Scheme 1 Chemical synthesis and structures of fentanyl (2c), fentalogues (2a, 2b, 2d–2k), heroin (HRN) and cocaine (COC). | ||
According to the U.S. Federal Control Substances Act 2017 published by the U.S. Department of Justice, fentanyl is classified as a Schedule II narcotic, since 1970. Recently, the Drug Enforcement Administration (DEA) in the U.S. provisionally listed structurally related fentanyl substances such as acetylfentanyl (2b) and butyrfentanyl (2f) under Schedule I of the Controlled Substances Act.6 Worldwide the fentanyl crisis has proliferated at an unprecedented rate posing significant public health challenges. In the U.S., 1013 fentanyl-related deaths were reported over the period 2005–2007, which were attributed to fentanyl/HRN mixtures. In March 2017, China controlled four types of fentanyl analogues under the Chinese law, after a meeting held between the U.S. In Europe, according to a commentary published in the International Journal of Drug Policy,7 Sweden was the first country to report 8 fentanyl-related deaths in 1994. However, Estonia recorded the highest fentanyl mortality rates in the EU, with an estimate of 1100 deaths over the period 2005–2013; deaths peaked in 2012 and reached 170, which was 11 times higher than the European average.7
Due to the high potency and dangerous impact of fentanyl and its analogues worldwide, their quantitative determination is of considerable importance. Within the literature, fentanyl is mainly determined via two major techniques, either chromatography (with the appropriate detector) or electrochemistry; Table 1 provides a thorough overview of these methodologies applied to the detection of fentanyl and its fentalogues. Chromatography and related techniques have extensively been reported but are generally considered expensive and require expert users for their efficient operation. In the case of electrochemical methodologies, these are more limited and are yet be utilized for the separation of HRN, fentanyl and fentalogues. Of note, from the inspection of Table 1, only one hybrid technique high-performance liquid chromatography-photolysis-electrochemical detection8 has been utilized towards fentanyl detection, however, only used for the analysis of 2c.
| Analytical method | Target analytes | Total run time (min) | Limit of detection (LOD)b | Limit of quantitation (LOQ)c | Linearity range | Ref. |
|---|---|---|---|---|---|---|
| a SPE-GC/MS: solid phase extraction-gas chromatography/mass spectrometry; UPLC-MS/MS: ultra-performance liquid chromatography tandem mass spectrometry; TD-DART-MS: thermal desorption-direct analysis in real time-mass spectrometry; IMS; ion mobility spectrometry; LC-MS/MS: liquid chromatography-tandem mass spectrometry; LC-MS: liquid chromatography-mass spectrometry; GC/MS: gas chromatography-mass spectrometry; PVC: polyvinyl chloride; GC: glassy carbon electrode; ASV: adsorptive stripping voltammetry; SDME: static dropping mercury electrode; HPLC-UV-EC: high-performance liquid chromatography-photolysis-electrochemical detection. b LOD is a range of the smallest and biggest LOD for the quantified analytes. c LOQ is a range of the smallest and biggest LOQ for the quantified analytes. d Δ = not disclosed. e LOD90 = corresponds to the lowest mass which can be detected of a wipe with a 90% probability of true detection. | ||||||
| SPE-GC/MSa | HRN and (2c) | 17 | 0.4 × 10−3 to 4.62 × 10−3 μg mL−1 | 1.3 × 10−3 to 14 × 10−3 μg mL−1 | 20 × 10−3 to 1500 × 10−3 μg mL−1 | 26 |
| UPLC-MS/MS | HRN and (2c) | 8 | 0.03–0.21 pg injected | 0.1–0.7 pg injected | 0.1–1000 pg injected | 27 |
| TD-DART-MS and IMS | HRN, (2b), (2c), (2e), (2h), (2j) and (2k) | Δd | 8.807 × 10−2 to 2.55 nge | Δ | 0.5–20 ng per wipe (TD-DART-MS) | 28 |
| 1.0–100 ng per wipe (IMS) | ||||||
| LC-MS/MS | HRN and (2c) | 20 | 8.0 × 10−5 to 2.0 × 10−3 μg mL−1 | 1.0 × 10−4 μg mL−1 | 1.0 × 10−4 to 7.60 × 10−1 μg mL−1 | 29 |
| LC-MS/MS | (2b), (2c), (2e), (2f), and (2j) | 13.5 | 0.017 × 10−3 to 0.056 × 10−3 mg mL−1 | 0.10 × 10−3 to 0.50 × 10−3 μg mL−1 | 0.1 × 10−3 to 50 × 10−3 μg mL−1 | 30 |
| LC-MS/MS | (2b) and (2c) | 5 | Δ | 0.1 × 10−3 to 1.0 × 10−3 μg mL−1 | 0.10 × 10−3 to 40 × 10−3 μg mL−1 | 31 |
| LC-MS | (2c) | Δ | 0.2 × 10−3 to 0.6 × 10−3 μg mL−1 | 0.6 × 10−3 to 2 × 10−3 μg mL−1 | Δ | 32 |
| GC/MS | (2c) | 23 | 0.08 × 10−3 μg mL−1 | 0.5 × 10−3 μg mL−1 | 0.5 × 10−3 to 50 × 10−3 μg mL−1 | 33 |
| HPLC utilizing absorbance rationing | (2c) | 20 | Qualitative analysis | Qualitative analysis | Qualitative analysis | 34 |
| Potentiometric method using PVC membrane electrode | (2c) | — | 1.83 μg mL−1 | Δ | 3.36–3.36 × 103 μg mL−1 | 35 |
| Potentiometric method using plastic membrane electrode (PVC) | (2c) | — | 2.11 μg mL−1 | Δ | 3.36–3.36 × 103 μg mL−1 | 36 |
| Electrochemiluminescence, cyclic voltammetry using GC electrode | (2c) | — | 2.86 × 10−3 μg mL−1 | Δ | 3.36 × 10−3 to 33.6 μg mL−1 | 37 |
| ASV using SDME | (2c) | — | 1.68 × 10−2 μg mL−1 | Δ | 3.36 × 10−2 to 3.36 × 10−1 μg mL−1 | 38 |
| Polarography | (2c) | — | 1.68 × 10−1 μg mL−1 | Δ | 3.36 × 10−1 to 6.73 μg mL−1 | 39 |
| HPLC-UV-EC | (2c) | Δ | Δ | Δ | 8 | |
| This work, HPLC-DAD (Method I) | HRN, (2a)–(2k) | 30 | HRN: 16.2 × 10−2 μg mL−1 | HRN: 54 × 10−2 μg mL−1 | 5.0–120 μg mL−1 | — |
| 2a: 22.49 × 10−2 μg mL−1 | 2a: 74.96 × 10−2 μg mL−1 | |||||
| 2b: 24.47 × 10−2 μg mL−1 | 2b: 81.57 × 10−2 μg mL−1 | |||||
| 2c: 41.67 × 10−2 μg mL−1 | 2c: 1.39 μg mL−1 | |||||
| 2d: 49.67 × 10−2 μg mL−1 | 2d: 1.66 μg mL−1 | |||||
| 2e: 68.80 × 10−2 μg mL−1 | 2e: 2.29 μg mL−1 | |||||
| 2f: 74.63 × 10−2 μg mL−1 | 2f: 2.49 μg mL−1 | |||||
| 2g: 95.54 × 10−2 μg mL−1 | 2g: 3.18 μg mL−1 | |||||
| 2h: 96.77 × 10−2 μg mL−1 | 2h: 3.23 μg mL−1 | |||||
| 2i: 1.15 μg mL−1 | 2i: 3.85 μg mL−1 | |||||
| 2j: 1.13 μg mL−1 | 2j: 4.5 μg mL−1 | |||||
| 2k: 1.36 μg mL−1 | 2k: 4.55 μg mL−1 | |||||
| This work, HPLC-AD (Method II) | 30 | HRN: 0.45 μg mL−1 | HRN: 1.49 μg mL−1 | 10.0–120 μg mL−1 | — | |
| 2a: 0.48 μg mL−1 | 2a: 1.61 μg mL−1 | |||||
| 2b: 0.48 μg mL−1 | 2b: 1.61 μg mL−1 | |||||
| 2c: 0.77 μg mL−1 | 2c: 2.58 μg mL−1 | |||||
| 2d: 0.91 μg mL−1 | 2d: 3.04 μg mL−1 | |||||
| 2e: 1.19 μg mL−1 | 2e: 3.95 μg mL−1 | |||||
| 2f: 1.28 μg mL−1 | 2f: 4.27 μg mL−1 | |||||
| 2g: 1.71 μg mL−1 | 2g: 5.71 μg mL−1 | |||||
| 2h: 1.68 μg mL−1 | 2h: 5.59 μg mL−1 | |||||
| 2i: 2.60 μg mL−1 | 2i: 8.68 μg mL−1 | |||||
| 2j: 2.78 μg mL−1 | 2j: 9.26 μg mL−1 | |||||
| 2k: 2.93 μg mL−1 | 2k: 9.76 μg mL−1 | |||||
Herein, we develop for the first time, a combination of high-performance liquid chromatography-diode array detection (HPLC-DAD) and high-performance liquid chromatography-amperometric detection (HPLC-AD) methods for the separation and quantification of fentanyl (2c), ten fentalogues (2a, 2b, 2d–2k) and HRN. This approach uses screen-printed graphite macroelectrodes (SPEs), as they are an advantageous electrochemical sensing platform that can be utilized in various quantification methods, owing to their economy of scales, simplicity and reproducibility.9–12
Results and discussion
Electrochemical measurement of the target analytes using cyclic voltammetry
Fig. 1 depicts the individual voltammetric profiles for fentanyl (2c), HRN and COC utilizing SPEs. In consideration of previous literature, fentanyl (2c)8,13 and cocaine (COC) (Scheme 1),14,15 as observed within Fig. 1, likely undergo an irreversible electrochemical oxidation/process as a result of the tertiary amine but in fact is likely more complex with the oxidation peak containing a contribution from more complex processes with formation of hydroxylated compounds and oxidation to give quinone imine groups. Since this is the first electrochemical report of the electrochemical oxidation of fentanyl, the exact mechanism needs further elucidation. Similarly, the observed electrochemical signature for heroin (HRN) is likely due to the irreversible electrochemical oxidation of the tertiary amine corresponding to the formation of a secondary amine.16,17 Fig. S1† displays typical cyclic voltammograms for each of HRN, COC, fentanyl and fentalogues, at pH 2.0, 7.0 and 10.0, utilizing SPEs. It is apparent that at pH 2.0 there are no apparent electrochemical oxidation waves/signal of the target analytes (Fig. S1A†) within the accessible electrochemical window. Additionally Fig. S1B and S1C† overview the response of the HRN, COC, fentanyl and fentalogues in pH 10 and 7; Table S1† compares the peak potentials of all the drug analytes at the pH's 2, 7 and 10. Note that analytically useful voltammetric peaks are obtained for all the analytes in pH 7.0; consequently, pH 7.0 was chosen as the optimum pH for the voltammetric analysis of the target analytes/drugs. In summary, it is clear that in consideration of the peak potentials of all the studied drugs/analytes, they all overlap, in terms of electrochemical oxidation potentials, so if they were all present in a real sample, such as in the case of a street sample with an unknown composition, as is typically the case, all analytes present within the sample cannot be unambiguously determined. Due to the inability of a direct electroanalytical approach to separate and quantify HRN, (2c) and fentalogues (2a, 2b, 2d–2k), attention was turned to the use of chromatography to separate and analyze the drugs of interest, using high-performance liquid chromatography with DAD and AD detection.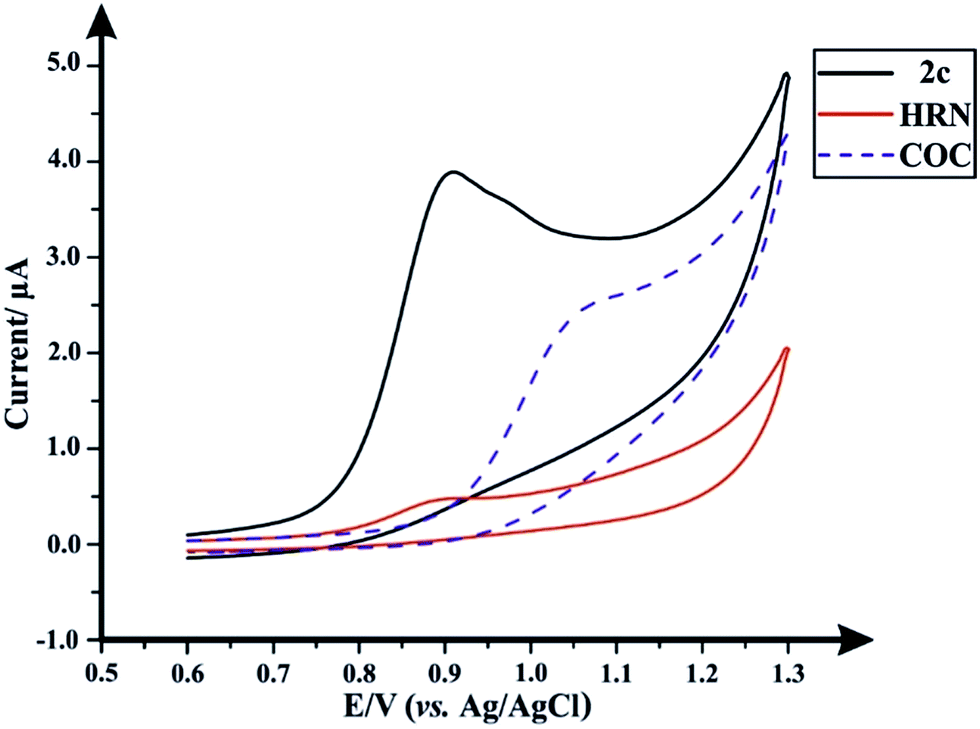 | ||
| Fig. 1 Typical cyclic voltammograms obtained separately for (2c), HRN and COC; solution composition: 50 μg mL−1 of each drug within a 0.04 M B–R buffer pH 7.0; scan rate: 50 mV s−1. | ||
Optimization of the chromatographic conditions
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :methanol 70:30% v/v) in combination with a suitable electrolyte (100 mM KCl).1 Similarly, we used a mobile phase consists of 20 mM ammonium formate:acetonitrile 70:30% v/v and added 100 mM KCl to the aqueous phase as an electrolyte. Attempts to decrease the organic modifier percentage were unfavorable as it affected the peaks symmetry and prolonged the run time over 30 minutes.
:methanol 70:30% v/v) in combination with a suitable electrolyte (100 mM KCl).1 Similarly, we used a mobile phase consists of 20 mM ammonium formate:acetonitrile 70:30% v/v and added 100 mM KCl to the aqueous phase as an electrolyte. Attempts to decrease the organic modifier percentage were unfavorable as it affected the peaks symmetry and prolonged the run time over 30 minutes.
Following the optimization of all the experimental/chromatographic conditions, typical HPLC-DAD chromatograms and HPLC-AD amperograms for HRN and fentalogues are presented in Fig. 2A and B, respectively. Fig. 2C and D represent HPLC-DAD chromatograms and HPLC-AD amperograms for COC, (2c) and fentalogues (2a, 2b, 2d–2k). It should be noted that HRN and COC elute at very similar retention times (tR) (COC; tR in HPLC-DAD = 2.65 min), therefore, analysis of HRN and COC in the same sample is likely not feasible. The proposed analytical methodologies were validated according to the ICH recommendations;21 all validation parameters are presented in Tables 2, 3, S2 and S3.†
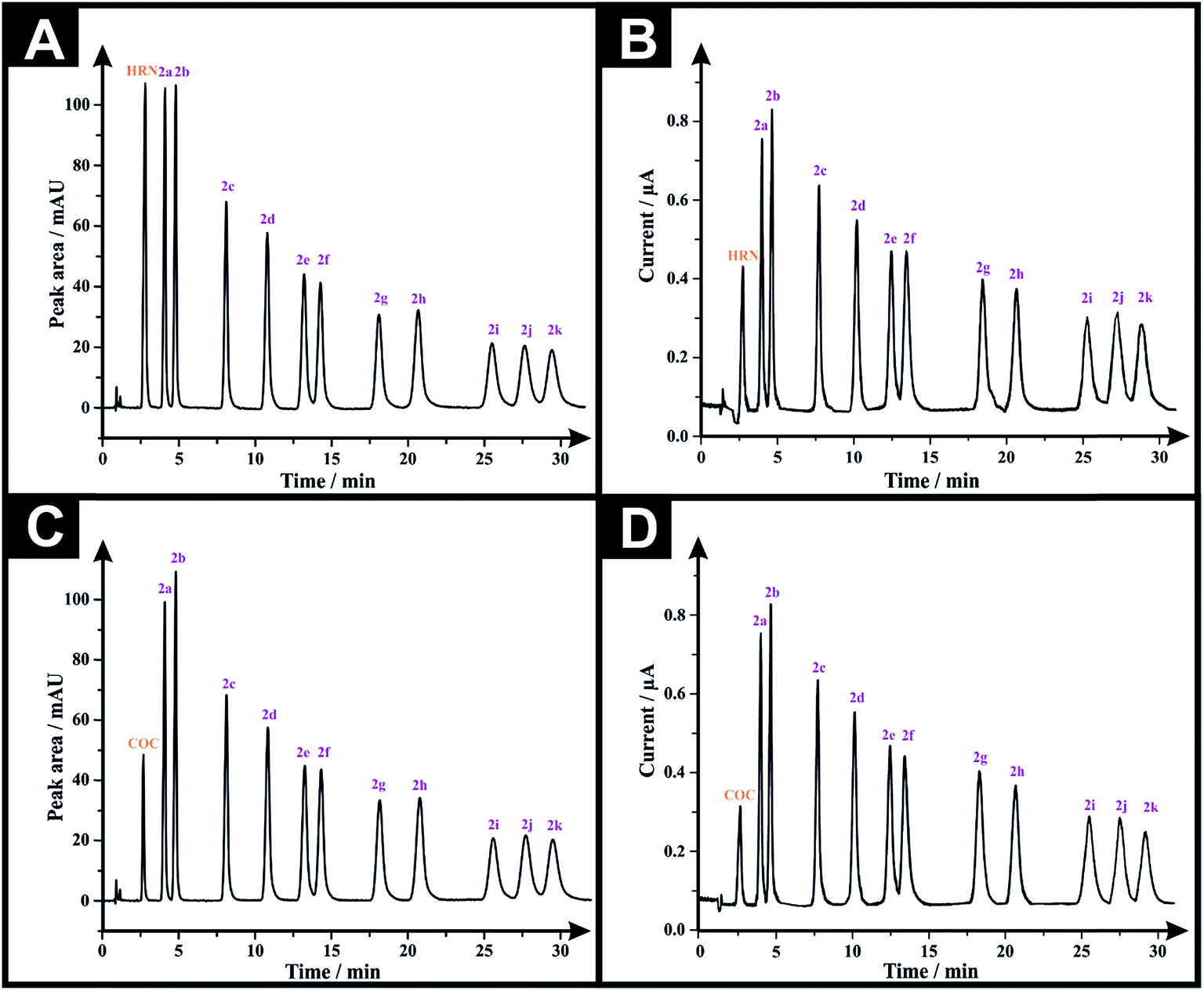 | ||
| Fig. 2 (A) Representative HPLC-DAD chromatograms for a solution containing 50 μg mL−1 of each of HRN, fentanyl (2c) and fentalogues (2a, 2b, 2d–2k) using an Eclipse XDB-C8 column (150 × 4.6 mm, i.d. 5 μm); mobile phase: acetonitrile:20 mM ammonium formate–100 mM potassium chloride buffer (pH 7.0) (30:70% v/v); flow rate 1.5 mL min−1, detector wavelength (UV): 205 nm and column temperature: 25 °C. (B) Representative amperogram for a solution containing 50 μg mL−1 of each of HRN, fentanyl and its 10 derivatives obtained using HPLC-AD flow cell system. (C) HPLC-DAD chromatogram for a solution containing 50 μg mL−1 of each of COC, fentanyl and its 10 derivatives. (D) Representative amperogram for a solution containing 50 μg mL−1 of each of COC, fentanyl and its 10 derivatives obtained using HPLC-AD flow cell system. | ||
:20 mM ammonium formate–100 mM potassium chloride buffer (pH 7.0) (30:70% v/v)]; flow rate = 1.5 mL min−1; detector wavelength (UV) = 205 nm; and column temperature = 25 °C
| Parameters | Analyte | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HRN | (2a) | (2b) | (2c) | (2d) | (2e) | (2f) | (2g) | (2h) | (2i) | (2j) | (2k) | |
| a R 2: co-efficient of regression. b y = 25.543x − 21.487. c y = 21.14x − 7.931. d y = 22.659x − 23.03. e y = 21.87x − 25.32. f y = 23.737x − 31.553. g y = 21.89x − 40.11. h y = 22.15x − 61.48. i y = 20.74x − 44.90. j y = 20.74x − 44.90. k y = 18.94x − 40.41. l y = 18.70x − 43.54. m y = 18.51x − 44.17. n LOD: limit of detection. o LOQ: limit of quantitation. p t R: retention time in minutes for drugs eluted from the chromatographic column (Method I). q RRT: relative retention time (determined with respect to (2c) retention time obtained from Method I). r k′: capacity factor. s N: number of theoretical plates expressed in plates per m. t HETP: height equivalent to theoretical plate expressed in m. u R s: resolution between two successive eluted peaks. v A s: asymmetry factor. w α: relative retention factor. | ||||||||||||
| R 2 | 0.999b | 0.999c | 0.999d | 0.999e | 0.999f | 0.999g | 0.999h | 0.999i | 0.999j | 0.999k | 0.999l | 0.999m |
| LODn (μg mL−1) | 16.20 × 10−2 | 22.49 × 10−2 | 24.47 × 10−2 | 41.67 × 10−2 | 49.67 × 10−2 | 68.80 × 10−2 | 74.63 × 10−2 | 95.54 × 10−2 | 96.77 × 10−2 | 1.15 | 1.35 | 1.36 |
| LOQo (μg mL−1) | 54.0 × 10−2 | 74.96 × 10−2 | 81.57 × 10−2 | 1.39 | 1.66 | 2.29 | 2.49 | 3.18 | 3.23 | 3.85 | 4.50 | 4.55 |
|
||||||||||||
| Precision (% RSD, n = 6) | ||||||||||||
| 5 μg mL−1 | 0.29 | 0.21 | 0.78 | 0.39 | 0.40 | 0.43 | 0.74 | 0.56 | 0.28 | 0.34 | 1.0 | 0.91 |
| 10 μg mL−1 | 0.42 | 0.42 | 0.94 | 0.86 | 0.64 | 0.42 | 1.0 | 1.0 | 0.75 | 0.53 | 0.55 | 0.53 |
| 40 μg mL−1 | 0.42 | 0.54 | 0.75 | 1.0 | 0.64 | 0.38 | 0.62 | 0.10 | 0.54 | 0.59 | 0.99 | 1.0 |
| 80 μg mL−1 | 0.90 | 0.77 | 0.66 | 0.55 | 0.61 | 0.83 | 0.74 | 0.55 | 0.58 | 0.55 | 0.46 | 0.46 |
| 120 μg mL−1 | 0.96 | 0.73 | 0.98 | 0.59 | 0.81 | 0.79 | 0.67 | 0.99 | 0.92 | 0.88 | 0.95 | 0.82 |
|
||||||||||||
| System suitability parameters | ||||||||||||
| t R ± SD (min)p (n = 6) | 2.79 ± 0.03 | 4.09 ± 0.04 | 4.79 ± 0.02 | 8.10 ± 0.04 | 10.79 ± 0.08 | 13.20 ± 0.05 | 14.26 ± 0.07 | 18.08 ± 0.01 | 20.65 ± 0.05 | 25.50 ± 0.03 | 27.64 ± 0.04 | 29.42 ± 0.06 |
| RRTq | 0.34 | 0.50 | 0.59 | 1.0 | 1.33 | 1.63 | 1.76 | 2.23 | 2.55 | 3.15 | 3.41 | 3.63 |
| k′r | 2.10 | 3.54 | 4.32 | 8.0 | 10.99 | 13.67 | 14.84 | 19.09 | 21.94 | 27.33 | 29.71 | 31.69 |
| N (plates)s | 2111 | 5165 | 6624 | 8396 | 8930 | 9238 | 9610 | 9563 | 9560 | 9853 | 10223 |
10196 |
| HETP (m)t | 7.11 × 10−5 | 2.90 × 10−5 | 2.26 × 10−5 | 1.79 × 10−5 | 1.68 × 10−5 | 1.62 × 10−5 | 1.56 × 10−5 | 1.57 × 10−5 | 1.57 × 10−5 | 1.52 × 10−5 | 1.47 × 10−5 | 1.47 × 10−5 |
| R s | — | 4.69 | 2.82 | 10.41 | 6.57 | 4.75 | 1.88 | 5.74 | 3.23 | 5.13 | 2.00 | 1.59 |
| A s | 1.29 | 1.43 | 1.33 | 1.00 | 0.92 | 0.91 | 0.88 | 0.82 | 0.88 | 0.80 | 0.93 | 0.84 |
| α | — | 1.69 | 1.22 | 1.85 | 1.37 | 1.24 | 1.09 | 1.29 | 1.15 | 1.25 | 1.09 | 1.07 |
Analytical performance was next considered via calibration curves for the HPLC-DAD and HPLC-AD methods. The LOD's (3-sigma) are found to range from 0.16–1.36 μg mL−1 and 0.45–2.93 μg mL−1, for HPLC-DAD and HPLC-AD, respectively (Tables 2 and 3). The LOQ's (10-sigma) range from 0.54–4.55 μg mL−1 and 1.49–9.76 μg mL−1, for HPLC-DAD and HPLC-AD, respectively (Tables 2 and 3). However, through comparing the LOD and LOQ of both analytical approaches we can conclude that HPLC-DAD is relatively more analytically sensitive than that of the HPLC-AD. All system suitability parameters are listed in Tables 2 and 3. The selectivity of both methods was investigated by injecting potential adulterants commonly found in street samples, which are: caffeine (Cf), paracetamol (Pc) and benzocaine (Bz). Unfortunately, the chromatographic approach could not separate (Cf) from (Pc), as the two peaks elute at similar retention times. Consequently, two adulterated solutions were prepared; one containing 50 μg mL−1 of each of (Cf), (Bz) and the twelve investigated drugs, while the second solution was the same but (Cf) was substituted with (Pc). Fig. 3A and B depict the HPLC-DAD chromatogram and HPLC-AD amperogram for the first adulterated solution and Fig. 3C and D present the HPLC-DAD chromatogram and HPLC-AD amperogram for the second adulterated solution, respectively. The HPLC-AD amperogram is presented within Fig. 3D, which depicts a peak for (Pc) and a peak for (Bz) while the HPLC-AD amperogram, illustrated in Fig. 3B, gives rise to a sole peak for (Bz). Such a response is due to (Cf) undergoing an irreversible electrochemical oxidation process, at Ep ≈ +1.4 V,22,23 however, herein, HPLC-AD measurements were conducted at Ep = +1.0 V (vs. Ag/AgCl) hindering the oxidation of (Cf). Finally, the specificity of both analytical methodologies was tested upon injecting a solution containing 300 μg mL−1 of potential diluents commonly found in pharmaceutical formulations (D-glucose, D-fructose, sucrose, lactose, starch, aerosil 200, sodium lauryl sulfate, stearic acid and sodium carboxymethyl cellulose). It was found that this solution did not show any peak in either HPLC-DAD chromatogram (Fig. S4A†) or HPLC-AD amperogram (Fig. S4B†). Also, a solution containing 300 μg mL−1 of excipients and 50 μg mL−1 of the investigated drugs was injected into the HPLC and the magnitude of the response (peak area and peak heights) of each compound was monitored for both methods (Fig. S5†). It was found that the added excipients did not interfere and did not affect the analytical sensing of the investigated drugs by both methods; the additives added were UV-inactive and electrochemically inert. This demonstrates the high specificity and selectivity of the promised sensing protocols.
:20 mM ammonium formate–100 mM potassium chloride buffer (pH 7.0) (30:70% v/v)]; flow rate = 1.5 mL min−1; detector wavelength (UV) = 205 nm; and column temperature = 25 °C
| Parameters | Analyte | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HRN | (2a) | (2b) | (2c) | (2d) | (2e) | (2f) | (2g) | (2h) | (2i) | (2j) | (2k) | |
| a R 2: co-efficient of regression. b y = 0.006x + 0.007. c y = 0.012x − 0.041. d y = 0.011x − 0.041. e y = 0.006x + 0.017. f y = 0.006x − 0.015. g y = 0.003x + 0.026. h y = 0.005x − 0.025. i y = 0.004x − 7 × 10−5. j y = 0.003x − 0.001. k y = 0.003x + 0.002. l y = 0.002x − 0.006. m y = 0.002x − 0.004. n LOD: limit of detection. o LOQ: limit of quantitation. p t R: retention time in minutes for drugs eluting from the flow-cell (Method II). q RRT: relative retention time (determined with respect to (2c) retention time obtained from Method II). r N: number of theoretical plates expressed in plates per m. s R s: resolution between two successive eluted peaks. | ||||||||||||
| R 2 | 0.997b | 0.996c | 0.996d | 0.999e | 0.997f | 0.996g | 0.998h | 0.999i | 0.998j | 0.996k | 0.999l | 0.998m |
| LODn (μg mL−1) | 0.45 | 0.48 | 0.48 | 0.77 | 0.91 | 1.19 | 1.28 | 1.71 | 1.68 | 2.60 | 2.78 | 2.93 |
| LOQo (μg mL−1) | 1.49 | 1.61 | 1.61 | 2.58 | 3.04 | 3.95 | 4.27 | 5.71 | 5.59 | 8.68 | 9.26 | 9.76 |
|
||||||||||||
| Precision (%RSD, n = 6) | ||||||||||||
| 10 μg mL−1 | 0.01 | 0.73 | 0.76 | 0.85 | 0.40 | 0.95 | 0.05 | 0.48 | 0.56 | 0.20 | 0.32 | 0.89 |
| 40 μg mL−1 | 0.10 | 0.57 | 0.06 | 0.55 | 0.38 | 0.13 | 0.49 | 0.81 | 0.16 | 0.41 | 0.79 | 0.06 |
| 60 μg mL−1 | 0.30 | 0.45 | 0.06 | 0.06 | 0.05 | 0.23 | 0.29 | 0.31 | 0.19 | 0.83 | 0.85 | 0.32 |
| 80 μg mL−1 | 0.20 | 0.15 | 0.09 | 0.06 | 0.13 | 0.11 | 0.08 | 0.33 | 0.11 | 0.20 | 0.77 | 0.06 |
| 100 μg mL−1 | 0.11 | 0.65 | 0.40 | 1.05 | 0.14 | 0.17 | 0.21 | 0.01 | 0.00 | 0.31 | 0.61 | 0.49 |
| 120 μg mL−1 | 0.92 | 0.84 | 0.33 | 0.63 | 0.27 | 0.60 | 0.82 | 0.21 | 0.31 | 0.93 | 0.96 | 0.32 |
|
||||||||||||
| System suitability parameters | ||||||||||||
| t R ± SD (min)p (n = 6) | 2.80 ± 0.03 | 4.1 ± 0.04 | 4.80 ± 0.02 | 8.11 ± 0.04 | 10.80 ± 0.08 | 13.21 ± 0.05 | 14.27 ± 0.07 | 18.09 ± 0.01 | 20.66 ± 0.05 | 25.51 ± 0.03 | 27.65 ± 0.04 | 29.43 ± 0.06 |
| RRTq | 0.35 | 0.51 | 0.59 | 1 | 1.33 | 1.63 | 1.76 | 2.23 | 2.55 | 3.15 | 3.41 | 3.63 |
| N (plates)r | 2101 | 5150 | 6620 | 8380 | 8920 | 9230 | 9600 | 9555 | 9545 | 9850 | 10218 |
10188 |
| R s | — | 4.69 | 2.82 | 10.41 | 6.57 | 4.75 | 1.86 | 5.74 | 3.18 | 5.10 | 1.88 | 1.50 |
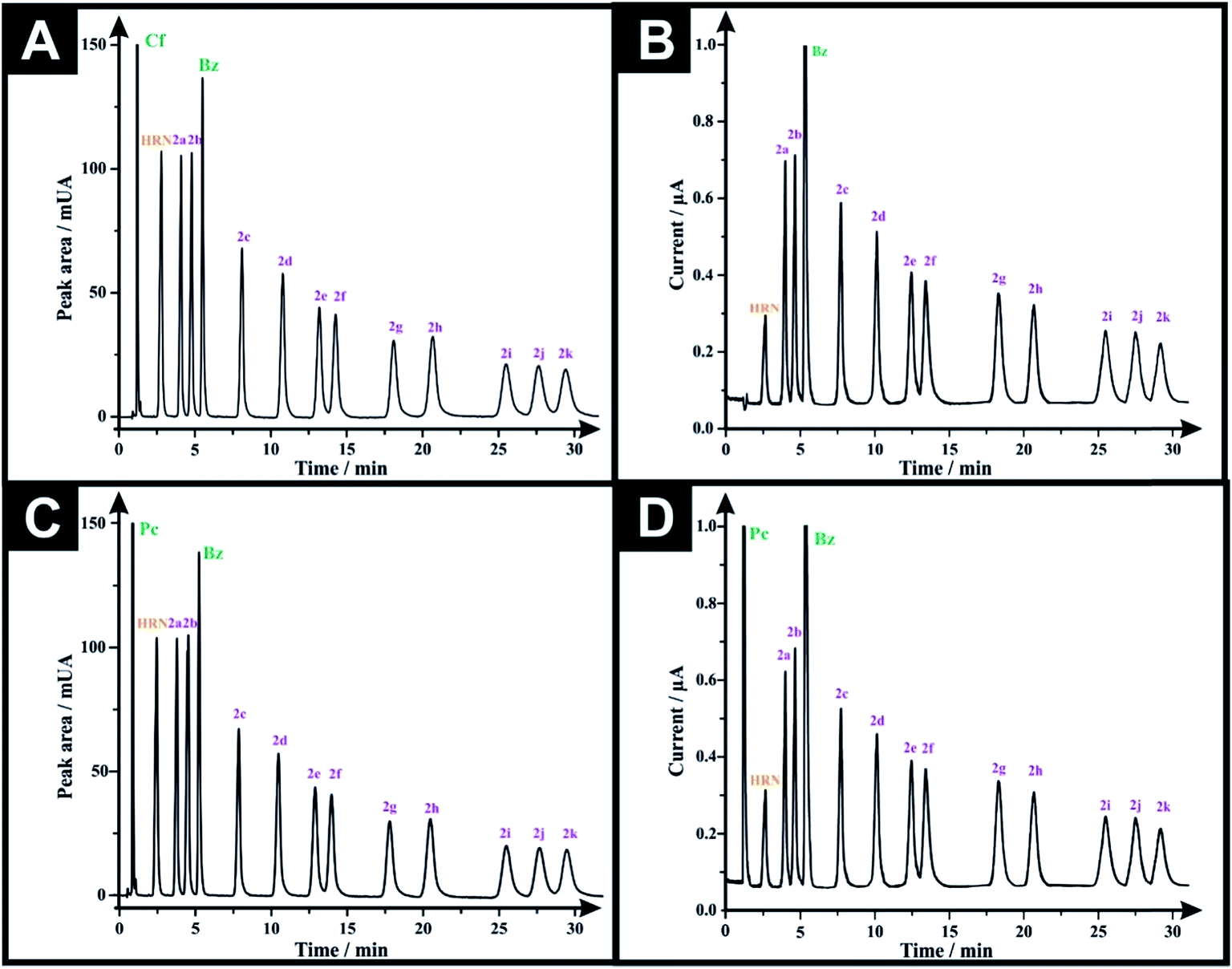 | ||
| Fig. 3 (A) Representative HPLC-DAD chromatograms for a solution containing 50 μg mL−1 of each of caffeine (Cf), HRN, benzocaine (Bz), fentanyl (2c) and its 10 derivatives using Eclipse XDB-C8 column (150 × 4.6 mm, i.d. 5 μm); mobile phase: acetonitrile:20 mM ammonium formate–100 mM potassium chloride buffer (pH 7.0) (30:70% v/v); flow rate 1.5 mL min−1, detector wavelength (UV): 205 nm and column temperature: 25 °C. (B) Representative amperogram for a solution containing 50 μg mL−1 of each of caffeine (Cf), HRN, benzocaine (Bz), fentanyl and its 10 derivatives obtained using HPLC-AD flow cell system. (C) HPLC-DAD chromatogram for a solution containing 50 μg mL−1 of each of paracetamol (Pc), HRN, benzocaine (Bz), fentanyl and fentalogues. (D) Representative amperogram for a solution containing 50 μg mL−1 of each of paracetamol (Pc), HRN, benzocaine (Bz), (2c) and fentalogues using HPLC-AD flow cell system. | ||
Three seized samples of suspected HRN, were provided under license by Greater Manchester Police and tested using our proposed HPLC-AD methodology in order to test the viability of the proposed protocol (as described in the Experimental section). As presented in Table 4, analysis of a 2 mg mL−1 HSS1 solution resulted in a % w/w equals to 1.66 ± 0.02 and 1.48 ± 0.02 for (2c), using HPLC-DAD and HPLC-AD, respectively (Table 4). Results for HRN% were strongly correlated with those obtained with GC-MS, where the obtained % w/w for HRN = 26.70 ± 0.61, however, the proposed methods overestimated (2c)% when compared with that of GC/MS, (2c)% = 0.26 ± 1.16 (Table 4). The results obtained by HPLC-DAD and HPLC-AD methodologies when analyzing HSS2, (containing HRN, (Cf) and (Pc)) and HSS3 (containing HRN only) were in a good agreement with each other: HRN % w/w in HSS2 = 29.09 ± 0.57 and 27.42 ± 0.27 using HPLC-DAD and HPLC-AD, respectively (Table 4); HSS3, HRN % w/w = 60.09 ± 0.36 and 58.92 ± 0.83 using HPLC-DAD and HPLC-AD, respectively (Table 4). When comparing those results with GC/MS, we find that HRN % w/w = 29.50 ± 0.52 and 62.70 ± 0.75 for HSS2 and HSS3, respectively, emphasizing the reliability of the proposed protocol. Note that HSS1 and HSS2 have (Pc) and (Cf), both adulterants, which elute together as a single peak using the HPLC-DAD, whereas, in HPLC-AD only the (Pc) peak was visible.
| Method of detection | Detected drugs of abuse | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HPLC-DAD (Method I) | HPLC-AD (Method II) | GC/MS | ||||||||||
| Analyzed street sample | HRN (% w/w) ± SDa | RSDb (%) | (2c) (% w/w) ± SDa | RSDb (%) | HRN (% w/w) ± SDa | RSDb (%) | (2c) (% w/w) ± SDa | RSDb (%) | HRN (% w/w) ± SDa | RSDb (%) | (2c) (% w/w) ± SDa | RSDb (%) |
| a Mean ± SD of obtained % w/w of three determinations of each detected drug in each street sample. b Percentage relative standard deviation of obtained % w/w of three determinations of each detected drug in each street sample. c n.d. = not detected. | ||||||||||||
| t R (min) ± SD | 2.79 ± 0.03 | 1.08 | 8.10 ± 0.05 | 0.62 | 2.80 ± 0.03 | 1.07 | 8.11 ± 0.05 | 0.62 | 12.75 ± 0.02 | 0.16 | 13.84 ± 0.003 | 0.02 |
| HSS1 | 26.64 ± 0.31 | 1.16 | 1.66 ± 0.02 | 1.20 | 24.16 ± 0.18 | 0.75 | 1.48 ± 0.02 | 1.35 | 26.70 ± 0.61 | 2.28 | 0.26 ± 0.003 | 1.15 |
| HSS2 | 29.09 ± 0.57 | 1.96 | n.d.c | n.d.c | 27.42 ± 0.27 | 0.98 | n.d.c | n.d.c | 29.50 ± 0.52 | 1.76 | n.d.c | n.d.c |
| HSS3 | 60.09 ± 0.36 | 0.60 | n.d.c | n.d.c | 58.92 ± 0.83 | 1.41 | n.d.c | n.d.c | 62.70 ± 0.75 | 1.20 | n.d.c | n.d.c |
In summary, in comparison of the two proposed detection protocols, there are no significant differences between them and both give rise to the reliable quantification of the target analytes. However, HPLC-AD demonstrates a slight underestimation in its values, which can be attributed to its relatively lower sensitivity comparing with the HPLC-DAD, but it is still suitable and sufficient for the quantification of heroin, fentanyl and fentalogues providing an analytically competitive protocol.
Conclusions
For the first time, the analytical determination of HRN, fentanyl (2c) and fentalogues (2a, 2b, 2d–2k) is reported using a dual HPLC-DAD and HPLC-AD protocol employing a commercially available impinging jet flow-cell, incorporating SPEs. This methodology demonstrates high accuracy, precision and is in a good agreement with traditional HPLC-DAD. This HPLC-AD technique is the first to be used as a precursor/screening method for forensic samples. In addition, this method was successfully applied for the analysis of the target analytes without interference of common formulated excipients or potential adulterants found in street samples.Experimental section
Chemicals and materials
All reagents are of commercial quality (Sigma-Aldrich, Gillingham, UK or Fluorochem Limited, Hadfield, UK) and used without further purification. All aqueous solutions were prepared with Milli-Q deionized water of resistively ≥ 18.2 Ω cm (Millipore system). All solutions (unless stated otherwise) were vigorously degassed, with highly pure nitrogen to remove oxygen prior to analysis. Solvents (Fisher Scientific, Loughborough, UK) were dried, where necessary, using standard procedures. The target compounds (2a–2k) were synthesized, from 4-ANPP (1), using an adaptation of the method reported by Valdez et al.24 and obtained as stable, off-white powders (Scheme 1). The hydrochloride salts were determined to be soluble (10 mg mL−1) in deionised water, methanol and dimethylsulfoxide. To ensure the authenticity of the materials utilized in this study the synthesized samples were structurally characterized (see ESI† for more details) by both 1H-NMR and 13C-NMR and the purity of all samples was confirmed by NMR and GC-MS (>99.5% in all cases). 1H-NMR and 13C-NMR spectra were acquired on a JEOL AS-400 (JEOL, Tokyo, Japan) NMR spectrometer operating at a proton resonance frequency of 400 MHz and referenced to the residual solvent peak (d6-DMSO: 1H-NMR δ = 2.50 ppm, 13C-NMR δ = 39.52 ppm). Three seized samples of heroin were provided by Greater Manchester Police, in accordance with license requirements and agreed procedures. Details of the street samples utilized are presented within the ESI.†Instrumentation
:20 mM ammonium formate–100 mM potassium chloride buffer (pH 7.0), 30:70% v/v] flowing at a rate of 1.5 mL min−1. The LC system was connected to a computer loaded with Agilent Chemstation (Ver. 10.02) software (Agilent Technologies, Wokingham, UK) for data analysis.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :20 mM ammonium formate–100 mM potassium chloride buffer (pH 7.0), 30:70% v/v].
The buffer system (20 mM ammonium formate–100 mM potassium chloride) was prepared into a 2.0 L volumetric flask by dissolving 2.52 g of ammonium formate and 14.91 g of potassium chloride in ultrapure deionized water, the pH was adjusted by the dropwise addition of 0.1 M NaOH to pH 7.0. Afterwards, appropriate proportions of each of the buffer and organic modifier were mixed to obtain a 2.0 L mobile phase of the desired ratio. Prior to use, the mobile phase was vacuum filtered through a 0.45 mm pore filter paper and degassed for 10 min at 25 °C using an ultrasonic bath.
:20 mM ammonium formate–100 mM potassium chloride buffer (pH 7.0), 30:70% v/v].
The buffer system (20 mM ammonium formate–100 mM potassium chloride) was prepared into a 2.0 L volumetric flask by dissolving 2.52 g of ammonium formate and 14.91 g of potassium chloride in ultrapure deionized water, the pH was adjusted by the dropwise addition of 0.1 M NaOH to pH 7.0. Afterwards, appropriate proportions of each of the buffer and organic modifier were mixed to obtain a 2.0 L mobile phase of the desired ratio. Prior to use, the mobile phase was vacuum filtered through a 0.45 mm pore filter paper and degassed for 10 min at 25 °C using an ultrasonic bath.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Newton-Mosharafa PhD Scholarship, funded by both the Egyptian Ministry of Higher Education and the British Council is thanked for supporting this research. Funding from the Engineering and Physical Science Research Council (Reference: EP/N001877/1) and a British Council Institutional Grant Link (No. 172726574) is also acknowledged.References
- P. W. Peng and A. N. Sandler, Anesthesiology, 1999, 90, 576–599 CrossRef CAS PubMed.
- M. LaBarbera and T. Wolfe, J. Psychoact. Drugs, 1983, 15, 293–301 CrossRef CAS PubMed.
- M. N. Pastore, Y. N. Kalia, M. Horstmann and M. S. Roberts, Br. J. Pharmacol., 2015, 172, 2179–2209 CrossRef CAS PubMed.
- M. I. Jumbelic, Am. J. Forensic Med. Pathol, 2010, 31, 18–21 CrossRef PubMed.
- D. A. Algren, C. P. Monteilh, M. Punja, J. G. Schier, M. Belson, B. R. Hepler, C. J. Schmidt, C. E. Miller, M. Patel, L. J. Paulozzi, M. Straetemans and C. Rubin, J. Med. Toxicol., 2013, 9, 106–115 CrossRef CAS PubMed.
- https://www.dea.gov/pr/multimedia-library/publications/drug_of_abuse.pdf, accessed 20 Jun 2018.
- J. Mounteney, I. Giraudon, G. Denissov and P. Griffiths, Int. J. Drug Pol., 2015, 26, 626–631 CrossRef PubMed.
- C. M. Selavka, I. S. Krull and I. S. Lurie, J. Chromatogr. Sci., 1985, 23, 499–508 CAS.
- K. Y. Zuway, J. P. Smith, C. W. Foster, N. Kapur, C. E. Banks and O. B. Sutcliffe, Analyst, 2015, 140, 6283–6294 RSC.
- L. R. Cumba, J. P. Smith, K. Y. Zuway, O. B. Sutcliffe, D. R. do Carmo and C. E. Banks, Anal. Methods, 2016, 8, 142–152 RSC.
- M. A. Mohamed, S. A. Atty, A. M. Yehia, C. W. Foster, C. E. Banks and N. K. Allam, ACS Omega, 2017, 2, 6628–6635 CrossRef CAS PubMed.
- M. A. Mohamed, D. M. El-Gendy, N. Ahmed, C. E. Banks and N. K. Allam, Biosens. Bioelectron., 2018, 101, 90–95 CrossRef CAS PubMed.
- C. M. Selavka and I. S. Krull, J. Liq. Chromatogr., 1987, 10, 345–375 CrossRef CAS.
- J. M. P. J. Garrido, M. P. M. Marques, A. M. S. Silva, T. R. A. Macedo, A. M. Oliveira-Brett and F. Borges, Anal. Bioanal. Chem., 2007, 388, 1799–1808 CrossRef CAS PubMed.
- M. de Jong, N. Sleegers, J. Kim, F. Van Durme, N. Samyn, J. Wang and K. De Wael, Chem. Sci., 2016, 7, 2364–2370 RSC.
- J. Garrido, C. Delerue-Matos, F. Borges, T. R. Macedo and A. Oliveira-Brett, Electroanalysis, 2004, 16, 1497–1502 CrossRef CAS.
- J. R. Barreira Rodriguez, V. Cabal Diaz, A. Costa Garcia and P. Tunon Blanco, Analyst, 1990, 115, 209–212 RSC.
- R. Guidelli, J. Electroanal. Chem. Interfacial Electrochem., 1971, 33, 291–302 CrossRef CAS.
- R. Guidelli, J. Electroanal. Chem. Interfacial Electrochem., 1971, 33, 303–317 CrossRef CAS.
- K. B. Oldham, J. Electroanal. Chem. Interfacial Electrochem., 1973, 41, 351–358 CrossRef CAS.
- ICH, Q2(R1), Validation of Analytical Procedures: Text and Methodology, International Conference on Harmonization, Geneva, 2005 Search PubMed.
- A. Câmpean, M. Tertiş and R. Săndulescu, Cent. Eur. J. Chem., 2011, 9, 466–473 CrossRef.
- B. C. Lourenção, R. A. Medeiros, R. C. Rocha-Filho, L. H. Mazo and O. Fatibello-Filho, Talanta, 2009, 78, 748–752 CrossRef PubMed.
- C. A. Valdez, R. N. Leif and B. P. Mayer, PLoS One, 2014, 9, e108250 CrossRef PubMed.
- J. P. Smith, J. P. Metters, C. Irving, O. B. Sutcliffe and C. E. Banks, Analyst, 2014, 139, 389–400 RSC.
- F. Bravo, D. Gonzalez and J. Benites, J. Chem. Soc., 2011, 56, 799–802 CAS.
- M. R. Boleda, M. T. Galceran and F. Ventura, J. Chromatogr. A, 2007, 1175, 38–48 CrossRef CAS PubMed.
- E. Sisco, J. Verkouteren, J. Staymates and J. Lawrence, Forensic Chem., 2017, 4, 108–115 CrossRef CAS PubMed.
- M. Gergov, P. Nokua, E. Vuori and I. Ojanperä, Forensic Sci. Int., 2009, 186, 36–43 CrossRef CAS PubMed.
- K. E. Strayer, H. M. Antonides, M. P. Juhascik, R. Daniulaityte and I. E. Sizemore, ACS Omega, 2018, 3, 514–523 CrossRef CAS PubMed.
- S. Sofalvi, H. E. Schueler, E. S. Lavins, C. K. Kaspar, I. T. Brooker, C. D. Mazzola, D. Dolinak, T. P. Gilson and S. Perch, J. Anal. Toxicol., 2017, 41, 473–483 CrossRef CAS PubMed.
- A. Skulska, M. Kala and A. Parczewski, Przegl. Lek., 2005, 62, 581–584 Search PubMed.
- S. Strano-Rossi, I. Alvarez, M. J. Tabernero, P. Cabarcos, P. Fernandez and A. M. Bermejo, J. Appl. Toxicol., 2011, 31, 649–654 CrossRef CAS.
- I. S. Lurie, A. C. Allen and H. J. Issaq, J. Liq. Chromatogr., 1984, 7, 463–473 CrossRef CAS.
- L. J. Peng, M. L. Wen and Y. Yao, J. Pharm. Biomed. Anal., 2002, 30, 667–673 CrossRef CAS PubMed.
- L. J. Peng, M. L. Wen and Y. Yao, Anal. Sci., 2001, 17, 815–818 CrossRef CAS PubMed.
- H. Dai, H. Xu, X. Wu, Y. Chi and G. Chen, Anal. Chim. Acta, 2009, 647, 60–65 CrossRef CAS PubMed.
- H. Guo, N. Hu and S. Lin, Talanta, 1994, 41, 1929–1932 CrossRef CAS PubMed.
- G. H. H. N. L. Shuchang, J. Beijing Norm. Univ., Nat. Sci., 1993, 3, 21 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ay00009g |
| This journal is © The Royal Society of Chemistry 2019 |