
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Using castor oil to separate microplastics from four different environmental matrices†
Thomas
Mani
 ,
Stefan
Frehland
,
Andreas
Kalberer
and
Patricia
Burkhardt-Holm
*
,
Stefan
Frehland
,
Andreas
Kalberer
and
Patricia
Burkhardt-Holm
*
Department of Environmental Sciences, The Man-Society-Environment Program, University of Basel, Vesalgasse 1, 4051 Basel, Switzerland. E-mail: patricia.holm@unibas.ch
First published on 25th February 2019
Abstract
The detection of environmental microplastics (MP) is limited by the need to rigorously separate polymers from the surrounding sample matrix. Searching for an affordable, low-risk and quick separation method, we developed a protocol to separate microplastics (size range: 0.3–1 mm; virgin polymers: PP, PS, PMMA and PET-G) from suspended surface solids (marine and fluvial) as well as soil and sediment using castor oil. We demonstrate effective separation of the four polymers in a spike-recovery experiment. The mean ± SD MP spike-recovery rate was 99 ± 4% with an average matrix reduction of 95 ± 4% (dry weight, n = 16). The protocol was validated by separating non-spiked environmental Rhine River suspended solids samples recovering 74 ± 13% of MP. There PS comprised 76% of the non-retrieved MP and additional H2O2 digestion was needed to sufficiently reduce the highly abundant natural matrix. This castor oil lipophilicity-based protocol (i) achieves high MP recovery rates as a function of its environmental matrix reduction ability and (ii) provides environmentally friendly, non-hazardous and resource-efficient separation of MP from four different, typically investigated environmental compartments using one and the same method. Based on the Rhine River sample validation, the protocol is a potent replacement for traditional density separation techniques. Samples with high biogenic concentrations may require additional digestion.
Introduction
Microplastics (MP; <5 mm) are an emerging contaminant with planetary boundary implications.1,2 MP have been widely reported in marine,3 freshwater4,5 and terrestrial6,7 environments, and their rising environmental concentrations, high bioavailability and ecotoxicological potential have led to escalating global concern regarding the effects of MP.8 Surveys on environmental MP are published nearly every week; however, vast inconsistencies prevail in the methodologies used for sampling, purification, quantification and chemical analysis.9 To minimise the interference of natural residues during chemical analysis (for instance Fourier Transform Infrared [FTIR] or Raman spectroscopy10), it is vital to rigorously separate synthetic polymers from the environmental matrix. A number of separation and purification processes are known in preparatory protocols for MP analysis, including electro-11 and density separation based on NaCl, ZnCl2 or NaI, and purification protocols using NaClO, HNO3, H2O2 or KOH.12,13 However, many of these techniques are complex, time consuming and require extensive sample manipulation, and are thus prone to contamination and loss.12Moreover, some reagents used for density separation or chemical purification of MP samples (e.g. ZnCl2,14 NaI,15 HCl,16 or H2O2 (ref. 17)) pose a threat to health and/or the environment if mismanaged. These risks may be of particular concern when laypersons employ such techniques, for example in nowadays emerging microplastics citizen science surveys.18–21 Apart from their ecological hazards, many of these reagents are costly, may consume precious resources that could be better used elsewhere, or – depending on the economic resources or geographical location of the investigating unit – may not be easily available.22 Due to the increasing, widespread demand for monitoring and risk assessment of environmental MP, simplification and democratisation of standardized methods for MP sampling, purification and quantification are urgently required.9,23 A generally applicable, efficient, accurate, rapid, cheap purification method could aid the compilation of valuable extensive datasets on the distribution of MP in different environments.
In this study, we aimed to develop a method that enables the separation of some of the most common types of MP particles found in the environment from a diverse set of typically investigated matrices. We based our approach on an oil extraction protocol for separating MP from sediments that employs canola oil.22 Here, we report a simple, rapid, cheap extraction protocol based on castor oil – which has a higher molecular weight than canola oil (933.45 vs. 876.6 g mol−1) – for efficient and accurate recovery of various MP particles (PP, PS, PMMA and PET-G) from four typical environmental sample matrices: fluvial suspended surface solids (FSS), marine suspended surface solids (MSS), marine beach sediments (MBS) and agricultural soil (AS). In order to validate the microplastic recovery as well as the matrix reduction potential on non-spiked environmental samples the castor oil separation protocol was executed on five Rhine River FSS samples. These were collected at different locations between Switzerland and the German–Dutch border in order to capture a variety of microplastics and biogenic residue abundance as well as diversity in polymers and state of polymer degradation.24,25
Methods
Environmental sample collection
Fluvial suspended surface solids (FSS) and marine suspended surface solids (MSS) were collected using a 0.3 mm neuston net mesh (see Table S1† for the details of all hereinafter mentioned materials and instruments) and marine beach sediments (MBS) and agricultural soil (AS) were collected using a stainless-steel spoon (Table S2, Fig. S1†). The samples were fractionated (0.3–1.0 mm for FSS and MSS and 0.063–1 mm for MBS and AS) using geological sieves and stored at 7 °C. Prior to analysis, all samples were dried at 40 °C for 24 h. Each environmental matrix was divided into four replicates with specific target dry weights to the nearest mg: 1.0 g for FSS and MSS and 10.0 g for MBS and AS. Due to formation of aggregates in the MSS and FSS after drying, these samples were disaggregated by adding 100 mL distilled water (aq. dest.) and stirring at 400 rpm and 60 °C for 15 min prior to the separation protocol. Five additional Rhine River FSS samples were collected to examine whether non-spiked field samples potentially containing MP could be efficiently separated using the oil separation protocol. The Rhine River samples were collected at different locations (Table S2†). Each sample was collected over 10 min from the centre of the river cross section using a Manta Trawl (mesh: 300 μm), resulting in a mean (±SD) filtered volume of 84.2 ± 8.7 m3 (ESI, Table S2†).Microplastics for spiking
The environmental samples were spiked with synthetic polymer particles to assess the MP recovery rate of the protocol. Selection of the polymers was based on (i) global production volumes26 and the frequency of reported identification in the environment27 and (ii) the inclusion of a range of polymer densities from below to above the specific density values of fresh water and saltwater.We used fragments of four common polymer types: polypropylene (PP; specific density, ρ = 0.84 g cm−1, Table S4, Fig. S5†), polystyrene (PS; 1.05 g cm−1, Table S5, Fig. S7†), polymethyl methacrylate (PMMA; 1.19 g cm−1, Table S6, Fig. S9†) and glycol modified polyethylene terephthalate (PET-G; 1.27 g cm−1, Table S7, Fig. S11†). PP, PS and PET are among the six most commonly produced plastics (including PE, PVC and PUR).28 PP and PS are typically two of the three most commonly identified polymers in environmental plastic studies (the other is PE).27 PMMA was additionally selected as a major representative MP identified in the Rhine River.24 Particle sizes ranged from 0.3–1 mm.
As the oil separation protocol involves an aqueous phase with a specific density (ρ) of ∼1 g cm−1, polymers that covered the specific density range of 0.84–1.27 g cm−1 were deliberately selected. This density-range coverage is also important for separation of MP from heavier matrices, such as MBS (ρ ∼ 2.6 g cm−1), as the aqueous phase of the water–matrix mixture in every separation process will have a specific density (ρ) of ∼1 g cm−1 after the heavier solid fraction has settled.
Particles were mechanically fragmented and sieved into small (0.3–0.5 mm) and large (0.5–1.0 mm) fractions (see Table S1† for material and instrument details). Each fraction was numerically quantified using a stereomicroscope and chemically analysed by attenuated total reflection (ATR)-FTIR. Spectra were compared against a reference spectra library using Opus 7.5 software.
Castor oil microplastic and environmental matrix separation protocol
For the four environmental matrices, the four replicates of each pre-weighed residue were transferred into a separation funnel (n = 16, see Table S1† for all material and instrument details), suspended in 100 mL aq. dest. water and spiked with 100 MP particles (15 small and 10 large fragments of each polymer type, resulting in n = 1600 MP particles for the entire experiment).The sealed funnels were shaken for 30 s by hand to ensure thorough mixing of the spiked samples. Ten mL of castor oil (Table S1†) was added to each replicate. To guarantee the entire sample made contact with the castor oil, the separation funnel was inverted and shaken for 1 min by hand. For this the separation funnel was firmly held using two hands at the top and the bottom, respectively, and vigorously shaken and rotated at shoulder level. Subsequently, the separation funnels were rotated back to their upright position and the walls and lid of the funnel were rinsed with 400 mL aq. dest. water to ensure any remaining residue and oil droplets were returned to the mixture. Thereafter, the MBS and AS samples were left to settle for 15 min and the MSS and FSS samples for 45 min, according to previous experience (please refer to Fig. S2† for a schematic diagram of the entire procedure).
The lower aqueous and solid phase was then drained from the separation funnel into a clean glass jar, sealed and stored at 7 °C. The remaining oil phase was drained, vacuum filtered onto ash-less hardened cotton/cellulose filter paper (pore size: 25 μm), and the filter was washed with 100 mL ethanol (EtOH, 96%). Before and during draining, the lid and walls of the funnel were thoroughly rinsed using an additional 100 mL EtOH to transfer all residue onto the filter. The filter paper was transferred to a glass Petri dish, sealed with parafilm and stored at 7 °C for visual polymer spike-recovery and further FTIR analysis.
For visual spike-recovery, the filters containing the separated oil fraction filtrates were dried at 40 °C for 24 h and weighed to define dw-reduction (in %, Table S3†). Finally, the extracted spiked polymer particles on the filter (Fig. S3†) were picked by hand, quantified using a stereomicroscope and then chemically analysed by FTIR (as described in the section “Microplastics for spiking”).
The five Rhine River FSS samples collected to examine non-spiked environmental microplastic recovery rates and matrix reduction efficiency (Table S2†) were subjected to the same castor oil separation protocol. Subsequently the separated sample fractions were rinsed with aq. dest. and dried. All resulting fractions (oil as well as the water and solid phase) were visually assessed for MP within the size range of 0.3–1 mm. Totally 978 putative environmental microplastic particles were detected in both the oil and water and solid phase of all five Rhine River samples combined, of which 40% were chemically investigated using FTIR. After the oil separation, further dw matrix reduction potential of these five FSS-samples was investigated by subjecting the oil-extracted, rinsed and dried fractions to H2O2. For this, the pre-weighed dry sample residues from the upper oil-phase were placed in separate glass Petri dishes (diameter 6 cm), covered with 10 mL of H2O2 (30%) and incubated at 50 °C for 18 hours (adapted from ref. 17). Subsequently, the sample residues were rinsed on a 300 μm mesh using aq. dest., re-transferred back to the Petri dishes, dried for 6.5 h at 60 °C and weighed (dw).
Quality control and protection against contamination
The effect of EtOH rinsing on preventing castor oil-FTIR interference22 was quantified. For this, spectral hit quality indices (HQI) of PS and PP MP (size range longest axis: 0.5–1.2 mm) were compared after four different treatments in triplicate: (i) untreated, pure MP, (ii) MP submerged in EtOH (96%) for 30 min, (iii) MP submerged in castor oil for 30 min and (iv) MP submerged in castor oil for 30 min and subsequently submerged in EtOH (96%) for 5 min. MP from treatment (iv) reached significantly higher HQIs compared to treatment (iii) (Fig. S13†). Interestingly, MP submerged in EtOH (ii) reached higher HQIs than untreated pure MP (i) (Fig. S13†), indicating a general benefit of EtOH treatment for MP spectroscopy, also outside the application of the presented protocol. To prevent samples from contamination, glassware was used whenever possible. Containers, such as Petri dishes, were always covered with a lid or aluminium foil when not in use. Where the use of plastic materials for processing was unavoidable (e.g. the PTFE stopcock in the separation funnel), the item was thoroughly rinsed before use with deionised water and EtOH (70%). White lab coats (100% cotton) were worn in the laboratory at all times. Nitrile gloves were worn whenever the operator's hands came into close contact with samples and glassware. To prevent cross-contamination between instruments or receptacles, all used items were thoroughly washed with warm water and labware detergent. Procedural blanks were run during the visual sample examination phase (∼4 h) to assess the laboratory atmosphere contamination potential (adapted from ref. 29). For this, three thoroughly rinsed glass Petri dishes (diameter: 13 cm) were placed uncovered on the laboratory bench during the entire visual sample examination phase. Subsequently they were rinsed and drained onto cotton/cellulose filter paper and the filter paper was visually examined under a super-lighted stereomicroscope. No MP fragments were recorded in any of the blanks.Statistical analysis
Statistical analyses were performed using GraphPad Prism 7.03 for Windows (GraphPad Software, La Jolla, CA, USA). A Kruskal–Wallis test was performed to evaluate differences between the four dw matrix reduction rates in the spike-recovery experiment. A Friedman test was run to assess differences of total microplastic recovery rates between the different matrices in the spike-recovery experiment. Both tests were followed by a Dunn's multiple comparison test to evaluate where differences lie. To compare matrix dw reduction of the five non-spiked Rhine River samples (i) after oil separation and (ii) after additional H2O2 treatment, a Kolmogorov–Smirnov test was applied. To compare HQIs of PS and PP particles after four different treatments as described in “Quality control and protection against contamination” unpaired t tests were carried out after a Shapiro–Wilk normality test.Results & discussion
Environmental matrix reduction and recovery of spiked microplastics
The oil separation protocol reduced the irrelevant part of the environmental matrices by a mean of 95 ± 4% (±SD, dw, n = 16). The highest matrix reduction was achieved for AS (98 ± 1%), followed by MBS (97 ± 1%), MSS (94 ± 1%) and FSS (91 ± 4%, n = 4 each). AS dw reduction was significantly higher than FSS (p < 0.01). The mean recovery rate for all four synthetic polymers (PP, PS, PMMA and PET-G) over all sample replicates was 99 ± 4%. Spiked MP with a large diameter (0.5–1 mm) were recovered at a rate of 100 ± 2%, and those with a small diameter (0.3–0.5 mm) were recovered at a rate of 98 ± 4%. The highest spike recovery rate was observed for the MSS samples, from which 100 ± 2% of spiked MP (of all polymer types and sizes) were recovered, followed by 99 ± 3% for the AS replicates, 99 ± 3% for the FSS replicates and 97 ± 5% for the MBS replicates. PP (both size fractions) had the highest recovery rate from all four environmental matrices (99 ± 3%), followed by PS (99 ± 3%), PMMA (99 ± 4%), and PET-G (98 ± 5%; Fig. 1, Table S3†).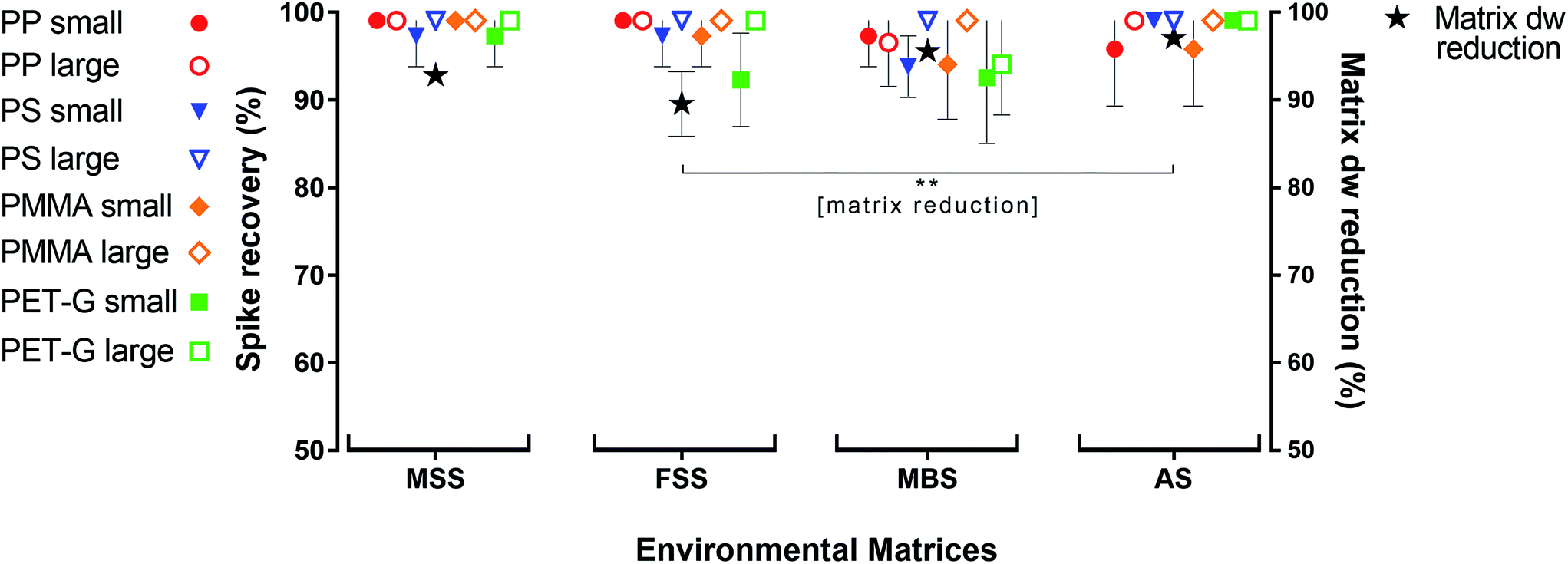 | ||
| Fig. 1 Mean matrix dry weight (dw) reduction and spike recovery rates (both in %) for the four polymer fragment types (PP, PS, PMMA and PET-G) from four environmental matrices: marine suspended surface solids (MSS), fluvial suspended surface solids (FFS), marine beach sediments (MBS), and agricultural soil (AS). Error bars indicate the standard deviation (n = 4 for every data point plotted). The small spiked polymer particles had a diameter of 0.3–0.5 mm (n = 15 per polymer and replicate), and the large spiked particles were 0.5–1 mm in diameter (n = 10 per polymer and replicate; resulting in a total of 4 × 25 MP particles = 100 spikes for each of the 16 experimental replicates). There was no significant difference in total microplastic recovery rates between tested environmental matrices (p > 0.05). The dw matrix reduction rates of FSS and AS samples differed significantly (** = p < 0.01). | ||
A before-and-after comparison using ATR-FTIR spectroscopy confirmed that the spiked polymers were not chemically altered during treatment (Fig. S6, S8, S10 and S12†). The non-destructive nature of the protocol is an important factor for bias-reduced environmental analysis; some published protocols involve potentially plastic-modifying steps such as acidic or alkaline purification30,31 or ultrasonication.32
Fluvial suspended solid samples (FSS) turned out to be the hardest to separate (Fig. 1), due to their high proportions of low-density biogenic particulate matter. Therefore, five non-spiked Rhine River FSS samples were subjected to the oil separation protocol for validation. Through the castor oil separation the environmental matrix could initially be reduced by 51 ± 11% dw. A subsequent H2O2 treatment of these remaining residues resulted in a significantly higher final dw matrix reduction of 82 ± 6% (Fig. 2). Clearing up the environmental matrix generally will also have a positive effect on the visual microplastic recovery rates, as sample insight improves on removal of non-plastic particles.13 We identified a total of 978 synthetic particles, distributed very heterogeneously among the five samples. This large range of MP concentrations represents the highly variable pollution levels of the different Rhine River stretches.24,25 Using the castor oil separation protocol, a mean 74 ± 13% of environmental MP could be retrieved in the upper oil phase (totally 773 MP particles retrieved from the upper oil phase, Fig. 2). Of the totally 205 MP particles retrieved from the lower aqueous and solid phase upon oil separation, the non-spiked Rhine River samples exhibited a large proportion of PS (76%, Fig. S4†). This was unexpected, as the overall recovery rate for PS particles in the spike-recovery experiment was 99%. PS opaque microbeads (33%) and PS foam (29%) were the largest contributors to the aqueous and solid PS abundance (Fig. S4†). The microbeads in the aqueous and solid phase (n = 67) stemmed from only one of the five environmental Rhine River samples. In an earlier study of the Rhine River such microbeads were identified to most likely be ion exchange resin (IER) beads.25 Possibly, the ion-active surface of IER beads weakens their lipophilicity, thus resulting in their relatively low recovery rate upon oil separation (67%). The other dominant shape-category found in the lower aqueous and solid phase was foams (68 of 205 MP, of which 60 were PS). Remarkably, despite their very low density (0.01–0.45 g cm−3 (ref. 33)) only 70% of foamed PS retained in the oil phase after separation. Possibly, their rough, scraggly surface promotes heteroaggregation with ambient, denser solid environmental particles and thus causes their settling below the oil phase.34 Nevertheless, the Rhine River sample findings demonstrate the applicability of the method to genuine field samples, albeit not quite yielding the same high level of recovery as in the spiking experiment. Among both the oil as well as the aqueous and solid phase of the Rhine River samples after separation, there was a high congruence of MP category abundance (solid fragments, foams, spherules, etc., Fig. S4†). However, the variety and abundance of different polymer types was distinctively poorer in the aqueous and solid phase (Fig. S4†). Fibres were present in both fractions but not accounted for in this investigation as their sound environmental and polymer identification is reportedly highly bias-afflicted.32,35 Due to the vast heterogeneity of MP abundance among the five Rhine River samples (11–692 MP) a statistical investigation of shape-related separation efficiency was not possible. Two samples with the highest MP abundances (692 and 235 MP) majorly influence the shape-related MP separation patterns depicted in Fig. S4.† This test reveals that weathered environmental MP, potentially clustered in compact heteroaggregates with non-plastic suspended solids, are not as well separable from the surrounding matrix as the spiked MP.
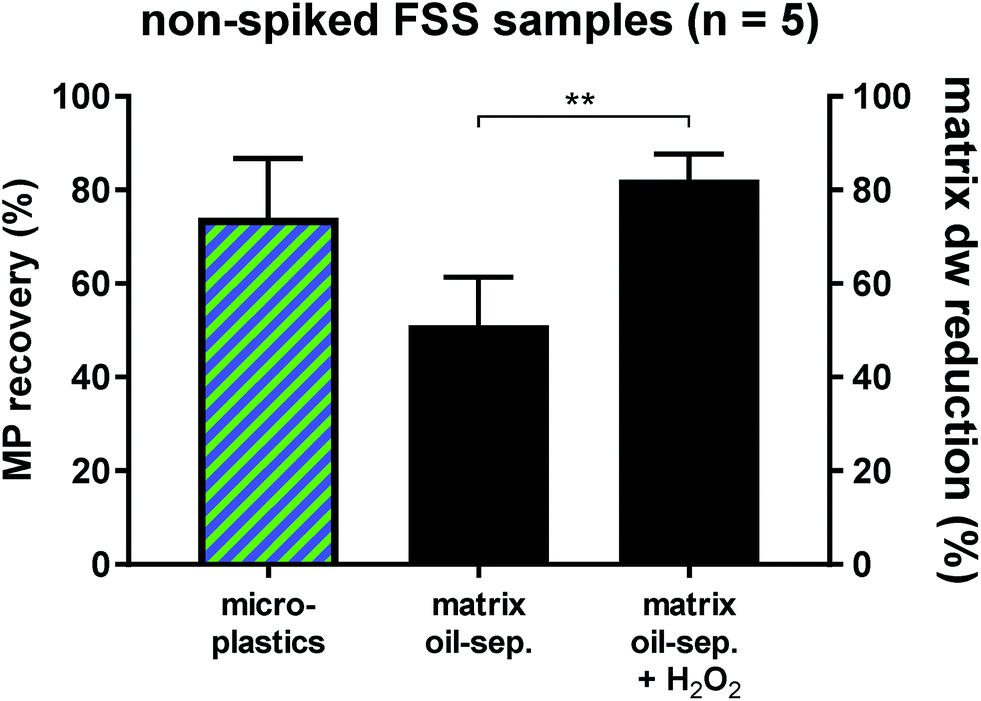 | ||
| Fig. 2 Mean percentage (+SD) of recovered microplastics (MP) from the five non-spiked Rhine River fluvial surface suspended solid samples (FSS, red and blue hatched column, left). The centre and right-hand columns show the mean percentage (+SD) of dw matrix reduction after the oil separation and after subsequent H2O2 treatment, respectively (n = 5). The dw matrix reduction was significantly higher after H2O2 treatment (** = p < 0.01). | ||
Characteristics and composition of the environmental matrices, such as FSS, may strongly vary depending on the season of collection, yielding varying abundance of e.g. lipophilic chitin crustacean exoskeletons.17 Furthermore, weathered, environmental MP are prone to alterations in their visual and chemical characteristics.36 Such potential modifications were apparent in the retrieved environmental microplastics in the form of e.g. fading colours and surface cracks. This finding alludes to the potential need for better disaggregation of environmental FSS samples prior to, and additional purification after oil separation.
Properties and advantages of a castor oil-based separation approach
In this study, we present a rapid, reliable method to extract commonly found MP with various polymeric characteristics from four typical environmental matrices, FSS, MSS, MBS and AS, that yields high polymer recovery and matrix reduction rates. In contrast to separation and purification protocols involving numerous treatment and sample transfer steps (e.g.ref. 17), the presented oil separation is non-toxic and performed practically entirely within a closed system, which is a great advantage in terms of reducing the risk of sample contamination. Furthermore, there is minimal necessity for sample transfer between containers during the protocol, and hence the risks of sample losses or contamination during transfer exposure are minimised. We show, however, that depending on the quality of the environmental matrix, additional purification after oil separation can be highly beneficial (e.g. using H2O2). Therefore, this oil separation protocol may, depending on the matrix at hand, serve as a valuable alternative to density separation, but not always as a replacement for further purification measures. Application of this single protocol to an array of distinct matrices fosters the potential for comparative research on MP pollution across different environmental compartments,9 while avoiding the need to use expensive and potentially hazardous reagents such as ZnCl2 (ref. 14) and NaI15 in density-based separation protocols or H2O2 (ref. 32) in enzymatic/oxidative purification protocols. The efficient isolation of any sample residue offered by the oil separation protocol represents an immensely important factor required for the success of both automatic and manual spectroscopic MP assessment techniques.17Previously reported separation techniques resulted in matrix reduction rates of up to 80% for MBS using fluidisation and flotation15 and 98% for MSS using enzymatic digestion.17 In comparison, the matrix reduction rate of this oil separation protocol (95 ± 4%) lies in the upper ranges of these other techniques. The presented castor oil separation protocol achieved very high MP spike-recoveries from four different matrices which are almost identical to an earlier published oil extraction protocol22 which was only tested on sediments (99 ± 4 vs. 99 ± 1.4%). Previous work on MBS using fluidisation and flotation reported spike recovery rates of between 18 and 100%37 and 91–99%.15 Spiked MP were recovered from MSS at a rate of 84 ± 3% using an enzymatic digestion protocol17 and 96–100% using the Munich Plastic Sediment Separator (MPSS).14 In comparison, the here presented protocol resulted in spike recovery rates of 99 ± 4% over all tested matrices.
Chemical considerations and background to the lipophilic castor oil approach
The natural castor oil employed in this protocol consists of approximately 99% long-chain C18 fatty acids (∼90% ricinoleic acid, C18H34O3).38 The high molecular weight of these long-chain aliphatic hydrocarbon-dominated fatty acids enables stable attraction between the non-polar lipophilic component of the fatty acid molecules and the non-polar lipophilic carbohydrate surface of synthetic polymer fragments (e.g. PP [C3H6]n) in a quasi-micellar manner. Furthermore, castor oil features one of the highest viscosities of the natural plant oils (>300 cP vs. <200 cP for canola oil), allowing the formation of a thick oil layer around the polymer fragments. The oil–polymer clusters have a lower overall density than water – even for high-density plastics such as PET-G (1.27 g cm−3). Therefore, these clusters move to the top of the separation funnel, where they merge with the castor oil and become separated from the lower aqueous environmental matrix phase. Due to the presence of a hydroxyl group on its twelfth C atom, ricinoleic acid is sufficiently polar to easily dissolve in EtOH following the oil separation procedure.In contrast to density-based separation approaches the here presented castor oil based microplastic separation protocol relies on the lipophilic and at the same time hydrophobic properties of synthetic hydrocarbon polymers.39,40 Within the separation funnel, separation and stratification of the liquid water/matrix and oil phases are driven by both chemical and gravitational forces. Hence, suspended solids with a specific density lower than water (∼1 g cm−3) but higher than castor oil (∼0.96 g cm−3) settle in the top layer of the water and solid phase, just below the oil phase. This phenomenon presents a challenge to precise manual separation of the water and oil phases while handling the polytetrafluorethylene (PTFE) stopcock during sample release for filtration. Especially during the separation of FSS, a large residue settled at the oil–water interface, which ultimately increased the total mass of solids oil-extracted from the matrix, thus limiting the matrix reduction rate for this matrix. In a previously published canola oil separation method, an additional enzymatic digestion step was applied when excess biomass was encountered during separation.22 Indicated by the strong reduction rates of the non-oleophilic matrix, as presented in our manuscript, it is most probable that highly biofouled MP, where contact between the castor oil and the polymer is inhibited, would not be separated as efficiently as unfouled MP. For samples where MP are strongly biofouled41,42 we would recommend applying sample digestion17 prior to oil separation to remove excess biogenic material from polymers. Further research is needed concerning the castor oil recovery potential specifically of biofouled, weathered, smaller (<0.3 mm) and denser MP (e.g. polytetrafluorethylene [PTFE] ∼ 2.2 g cm−3 (ref. 43)). We suggest repeating the separation process for the lower lying water–matrix phase in a series of further separation run-throughs. In our hands, a series of further separation run-throughs led to further separation and enhanced the rates of recovery, but residue reduction and MP recovery clearly depended on the environmental matrix and the characteristics of the MP particles (such as size, tendency to form aggregates, etc.).
Conclusions
Still today, after more than a decade of intensive research on microplastics worldwide, at least two major handicaps prevail: (i) there is a downright lack of uniformity in sampling, processing and analysis within the scientific community, and (ii) the inevitable separation and purification of environmental samples prior to the identification and analysis of potential plastics is more often than not enormously time and material consuming as well as prone to sample manipulation. Every methodology (e.g. oil-, density-, electro- or visual separation) ultimately quantifies a spectrum of the possible variables. Besides developing uniform protocols to guarantee comparability of environmental data, knowing the limits of each method is crucial, as it will facilitate to identify the most appropriate approach for every given case. Here, we were able to present a separation protocol for microplastics from environmental matrices which is highly simple and extremely efficient regarding the investments of time, material resources and health/environmental risk. The very same procedure was successfully demonstrated on four different types of environmental samples from the hydro- and lithosphere where the anthroposphere overlaps or affects them. This advance could possibly lead to a break-through in improving methodical homogeneity across the field and accelerate the accumulation of ever so important data for moving to the next crucial steps in microplastics sciences – namely, evaluating their ecological impact and finding mitigation and solution measures.Conflicts of interest
No competing financial interests exist.Acknowledgements
The authors gratefully acknowledge the officers and crew of the Swiss police vessels in Brugg and Basel, the Spanish vessel FIRMM Vision in Tarifa, as well as the German Waterway and Shipping Administration (WSA) vessels VSS Köln in Bad Honnef and VSS Grieth in Rees for their generous support in collecting the environmental samples. We would also like to extend our appreciation to Prof. Dr Luiz Felippe De Alencastro and his team from the Swiss Federal Institute of Technology Lausanne, for the loan of a Manta Trawl. Warm gratitude for impeccable laboratory guidance is expressed towards the technical assistants Nicole Seiler-Kurth and Heidi Schiffer at the Man-Society-Environment Program, Department of Environmental Sciences, University of Basel. We would also like to forward our gratitude to the reviewers for critically evaluating our initial manuscript and providing input that substantially enhanced the quality of this work. And finally, very special thanks go to Andrea Devlin, PhD, chief editor of Science Editing Experts, for her immaculate and fast proof reading and language support. This study was partially funded by theWorld Wide Fund for Nature (WWF) Switzerland.References
- A. Jahnke, H. P. H. Arp, B. I. Escher, B. Gewert, E. Gorokhova, D. Kühnel, M. Ogonowski, A. Potthoff, C. Rummel, M. Schmitt-Jansen, E. Toorman and M. MacLeod, Environ. Sci. Technol. Lett., 2017, 4, 85–90 CrossRef CAS.
- D. Sedlak, Environ. Sci. Technol., 2017, 51, 7747–7748 CrossRef CAS PubMed.
- M. Cole, P. Lindeque, C. Halsband and T. S. Galloway, Mar. Pollut. Bull., 2011, 62, 2588–2597 CrossRef CAS PubMed.
- D. Eerkes-Medrano, R. C. Thompson and D. C. Aldridge, Water Res., 2015, 75, 63–82 CrossRef CAS PubMed.
- M. O. Rodrigues, N. Abrantes, F. J. M. Gonçalves, H. Nogueira, J. C. Marques and A. M. M. Gonçalves, Sci. Total Environ., 2018, 633, 1549–1559 CrossRef CAS PubMed.
- M. C. Rillig, Environ. Sci. Technol., 2012, 46, 6453–6454 CrossRef CAS PubMed.
- G. S. Zhang and Y. F. Liu, Sci. Total Environ., 2018, 642, 12–20 CrossRef CAS PubMed.
- GESAMP, Sources, fate and effects of microplastics in the marine environment: part two of a global assessment, GESAMP, vol. 93, 2016 Search PubMed.
- BASEMAN, Interdisciplinary Research for Good Environmental Status, available at: http://www.jpi-oceans.eu/baseman, accessed 9 May 2018.
- A. Käppler, D. Fischer, S. Oberbeckmann, G. Schernewski, M. Labrenz, K.-J. Eichhorn and B. Voit, Anal. Bioanal. Chem., 2016, 408, 8377–8391 CrossRef PubMed.
- S. Felsing, C. Kochleus, S. Buchinger, N. Brennholt, F. Stock and G. Reifferscheid, Environ. Pollut., 2018, 234, 20–28 CrossRef CAS PubMed.
- A. B. Silva, A. S. Bastos, C. I. L. Justino, J. P. da Costa, A. C. Duarte and T. A. P. Rocha-Santos, Anal. Chim. Acta, 2018, 1017, 1–19 CrossRef CAS PubMed.
- N. P. Ivleva, A. C. Wiesheu and R. Niessner, Angew. Chem., 2017, 56, 1720–1739 CrossRef CAS PubMed.
- H. K. Imhof, J. Schmid, R. Niessner, N. P. Ivleva and C. Laforsch, Limnol. Oceanogr.: Methods, 2012, 10, 524–537 CrossRef CAS.
- M.-T. Nuelle, J. H. Dekiff, D. Remy and E. Fries, Environ. Pollut., 2014, 184, 161–169 CrossRef CAS PubMed.
- M. Cole, H. Webb, P. K. Lindeque, E. S. Fileman, C. Halsband and T. S. Galloway, Sci. Rep., 2014, 4, 4528 CrossRef PubMed.
- M. G. J. Löder, H. K. Imhof, M. Ladehoff, L. A. Löschel, C. Lorenz, S. Mintenig, S. Piehl, S. Primpke, I. Schrank, C. Laforsch and G. Gerdts, Environ. Sci. Technol., 2017, 51, 14283–14292 CrossRef PubMed.
- M. Bergmann, B. Lutz, M. B. Tekman and L. Gutow, Mar. Pollut. Bull., 2017, 125, 535–540 CrossRef CAS PubMed.
- T. Bosker, P. Behrens and M. G. Vijver, Integr. Environ. Assess. Manage., 2017, 13, 536–541 CrossRef PubMed.
- B. Gewert, M. Ogonowski, A. Barth and M. MacLeod, Mar. Pollut. Bull., 2017, 120, 292–302 CrossRef CAS PubMed.
- F. A. E. Lots, P. Behrens, M. G. Vijver, A. A. Horton and T. Bosker, Mar. Pollut. Bull., 2017, 123, 219–226 CrossRef CAS PubMed.
- E. M. Crichton, M. Noël, E. A. Gies and P. S. Ross, Anal. Methods, 2017, 9, 1419–1428 RSC.
- European Union, Strategy for plastics in a circular economy, 2018, http://https://ec.europa.eu/commission/publications/documents-strategy-plastics-circular-economy_en, accessed 10 October 2018 Search PubMed.
- T. Mani, A. Hauk, U. Walter and P. Burkhardt-Holm, Sci. Rep., 2015, 5, 17988 CrossRef CAS PubMed.
- T. Mani, P. Blarer, F. R. Storck, M. Pittroff, T. Wernicke and P. Burkhardt-Holm, Environ. Pollut., 2018, 245, 634–641 CrossRef PubMed.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- S. H. Hong, W. J. Shim and L. Hong, Anal. Methods, 2017, 9, 1361–1368 RSC.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- M. Bergmann, V. Wirzberger, T. Krumpen, C. Lorenz, S. Primpke, M. B. Tekman and G. Gerdts, Environ. Sci. Technol., 2017, 51, 11000–11010 CrossRef CAS PubMed.
- M. Claessens, L. van Cauwenberghe, M. B. Vandegehuchte and C. R. Janssen, Mar. Pollut. Bull., 2013, 70, 227–233 CrossRef CAS PubMed.
- G. Liebezeit and F. Dubaish, Bull. Environ. Contam. Toxicol., 2012, 89, 213–217 CrossRef CAS PubMed.
- M. G. J. Löder and G. Gerdts, in Marine Anthropogenic Litter, ed. M. Klages, L. Gutow and M. Bergmann, Springer, Cham, Heidelberg, New York, Dordrecht, London, 2015, pp. 201–227 Search PubMed.
- H. Winterling and N. Sonntag, Kunstst. Int., 2011, 10, 18–22 Search PubMed.
- M. Kooi, E. Besseling, C. Kroeze, A. P. van Wezel and A. A. Koelmans, in Freshwater microplastics. Emerging environmental contaminants?, ed. M. Wagner, S. Lambert, E. Besseling and F. J. Biginagwa, Springer Open, Cham, 2018, pp. 125–152 Search PubMed.
- V. Hidalgo-Ruz, L. Gutow, R. C. Thompson and M. Thiel, Environ. Sci. Technol., 2012, 46, 3060–3075 CrossRef CAS PubMed.
- A. Ter Halle, L. Ladirat, X. Gendre, D. Goudouneche, C. Pusineri, C. Routaboul, C. Tenailleau, B. Duployer and E. Perez, Environ. Sci. Technol., 2016, 50, 5668–5675 CrossRef CAS PubMed.
- A. Stolte, S. Forster, G. Gerdts and H. Schubert, Mar. Pollut. Bull., 2015, 99, 216–229 CrossRef CAS PubMed.
- J. Salimon, D. A. M. Noor, A. T. Nazrizawati, M. Y. M. Firdaus and A. Noraishah, Sains Malays., 2010, 39, 761–764 CAS.
- M. R. Rice and H. S. Gold, Anal. Chem., 1984, 56, 1436–1440 CrossRef CAS.
- Y. Mato, T. Isobe, H. Takada, H. Kanehiro, C. Ohtake and T. Kaminuma, Environ. Sci. Technol., 2001, 35, 318–324 CrossRef CAS PubMed.
- E. R. Zettler, T. J. Mincer and L. A. Amaral-Zettler, Environ. Sci. Technol., 2013, 47, 7137–7146 CrossRef CAS PubMed.
- I. V. Kirstein, S. Kirmizi, A. Wichels, A. Garin-Fernandez, R. Erler, M. Löder and G. Gerdts, Mar. Environ. Res., 2016, 120, 1–8 CrossRef CAS PubMed.
- AFT Fluorotec, The Properties and Advantages of PTFE, available at: https://www.fluorotec.com/news/blog/the-properties-and-advantages-of-polytetrafluoroethylene-ptfe/, accessed 21 February 2019.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ay02559b |
| This journal is © The Royal Society of Chemistry 2019 |