
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChiral micellar electrokinetic chromatographic separation for determination of L- and D-primary amines released from murine islets of Langerhans†
Kimberly
Evans
 ,
Xue
Wang
and
Michael G.
Roper
*
,
Xue
Wang
and
Michael G.
Roper
*
Department of Chemistry and Biochemistry, Florida State University, 95 Chieftan Way, Dittmer Building, Tallahassee, FL 32306, USA. E-mail: roper@chem.fsu.edu; Tel: +1-850-644-1846
First published on 18th February 2019
Abstract
D-Amino acids have been located in various tissues including the endocrine portion of the pancreas, the islets of Langerhans. D-Serine (D-Ser) is of particular interest since it is an agonist for the ionotropic N-methyl-D-aspartate receptors. To examine the potential release of D-Ser and other D-amino acids from islets, a chiral micellar electrokinetic chromatography method was developed by derivatizing primary amines with 2,3-naphthalenedicarboxaldehyde and to achieve resolution of the enantiomers, two surfactants were used in the separation, sodium dodecyl sulfate and sodium deoxycholate. With the optimized conditions, 36 small molecule standards, including four internal standards, were evaluated. For the 17 compounds that were fully resolved, limits of detection were less than 10 nM. The resulting optimized separation method produced high efficiency peaks, with an average of 300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 theoretical plates per peak and a peak capacity of 120. The method was used to examine the release of small molecules from groups of 50 murine islets of Langerhans. A peak was detected from islets incubated with 20 mM glucose that co-migrated with a D-Ser standard, although its level was below the quantifiable limit.
000 theoretical plates per peak and a peak capacity of 120. The method was used to examine the release of small molecules from groups of 50 murine islets of Langerhans. A peak was detected from islets incubated with 20 mM glucose that co-migrated with a D-Ser standard, although its level was below the quantifiable limit.
Introduction
Type 2 diabetes mellitus is a metabolic disorder that has become a pandemic. Each year, larger numbers of people are diagnosed with this disease, with global numbers approaching 422 million in 2014 alone.1 A common trait among these individuals is their inability to produce enough insulin to counteract the detrimental effects of high glucose. Unfortunately, although the effects of pancreatic hormones, such as insulin, released from islets of Langerhans on glucose regulation are fairly well-known, the mechanisms leading to this disease are still not completely understood.2Islets of Langerhans are comprised of multiple cell types, each secreting different peptide hormones.3 β-Cells, the most predominant of the cell types, release insulin which signals the body to store excess glucose. α-Cells, the next most predominant cell type, secrete glucagon to signal for initiation of glycogenolysis and gluconeogenesis when blood glucose levels are low. In diabetes, the release of these peptide hormones is dysregulated, leading to uncontrolled glucose levels. With recent findings that islets also release small molecules that can affect intra-islet or inter-organ communication,4–7 understanding how the release of these small molecules change from normal to diseased state is gaining interest for potential therapeutic intervention.
Islets release a number of small molecules that are traditionally classified as neurotransmitters, such as γ-aminobutyric acid (GABA), adenosine triphosphate (ATP), glycine (Gly), and glutamate (Glu).4–12 The release of these have been hypothesized as signalling molecules for communication between islet cell types, islets and the nervous system, or as substrates for various metabolic pathways. While these classical neurotransmitters are released from islets, it is uncertain if the release of D-amino acids (DAA), also known to act as neurotransmitters/neuromodulators, play a role in islet biology.
D-Alanine (Ala), D-aspartate (Asp), D-Glu, and D-Ser have been detected in pancreatic tissue, although not all have been localized to islets.13–18 Of the DAA mentioned, D-Asp and D-Ala are the only DAA to have been detected14 and quantified15 in islet lysate and secretions. D-Ser, an agonist for N-methyl-D-aspartate receptors (NMDAR), has been detected in a variety of rodent tissue, including the pancreas,16–18 but to the best of our knowledge, has not been localized to islets.
NMDAR activation requires binding by both glutamate and either Gly or D-Ser in addition to membrane depolarization.19,20 Opening of these channels allows flux of cations through the membrane, and can also affect flux of ions through other ion channels.21 The resulting ionic flux can affect the overall membrane potential and lead to different cellular outcomes depending on what is activated.21 The presence of these receptors have been documented on rodent cell lines and islets,22,23 and human islets,21 and although their role has yet to be defined, studies on inhibiting these receptors showed promising results for enhanced glucose-stimulated insulin secretion and islet survival.21 While either Gly or D-Ser can be a ligand for the receptor,19,20 ser racemase (Srr), the enzyme responsible for conversion of L-Ser to D-Ser, was found in both human and murine islets.24 Together, the presence of both Srr and NMDAR in murine and human islets raises the possibility that release of D-Ser from islets may play a role in autocrine or paracrine signaling through NMDAR within the islet or to nearby synaptic terminals, similar to other small molecules released from in islets.4–11 Direct drug targeting of these receptors could be part of a step forward in better managing diabetes for people worldwide.
In this study, we developed a chiral micellar electrokinetic chromatography (MEKC) method that enabled separation of several DAA and other primary amines released from murine islets of Langerhans. Limits of detection for those that were fully resolved were less than 10 nM with linearity greater than 0.99 by linear regression. The critical pair of D- and L-Ser had resolution of 1.7 and LOD values of 4 nM. Using this method, release from a group of 50 murine islets incubated at different glucose conditions were examined. A peak was observed in the high glucose sample that co-migrated with D-Ser standard spikes; however, its concentration in these samples was below the quantifiable limit, which precluded our ability to confidently identify this compound as D-Ser.
Methods and materials
Chemicals and reagents
Sodium tetraborate, dextrose, and 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES) were from Fisher Scientific (Pittsburg, PA). Cosmic Calf Serum was from HyClone Laboratories (South Logan, UT). Sodium hydroxide (NaOH) and boric acid were purchased from EMD chemicals (San Diego, CA). RPMI 1640 was from Mediatech (Manassas, Va). Acetonitrile (ACN) was from Avantor Performance Materials (Center Valley, PA). Gentamicin was from Lonza (Walkersville, MD). Collagenase P was purchased from Roche Diagnostics (Indianapolis, IN). All other chemicals were from Sigma-Aldrich (St. Louis, MO) unless otherwise noted. All solutions were made with ultrapure DI water (NANOpure® Diamond system, Barnstead International, Dubuque, IA) and filtered with 0.2 μm nylon syringe filters from Pall Corporation (Port Washington, NY) unless otherwise noted. The pH for all solutions were adjusted using 1 or 5 M NaOH as needed.Amino acid derivatization
Stock solutions of L-Ser, D-Ser, L-threonine (L-Thr), D-Thr, L-asparagine (L-Asn), L-glutamine (L-Gln), D-Gln, L-Glu, D-Glu, L-Ala, D-Ala, L-histidine (L-His), L-Asp, D-Asp, L-tyrosine (L-Tyr), D-Tyr, GABA, L-valine (L-Val), D-Val, L-methionine (L-Met), D-Met, L-isoleucine (L-Ile), L-leucine (L-Leu), D-Leu, L-phenylalanine (L-Phe), D-Phe, L-tryptophan (L-Trp), D-Trp, L-arginine (L-Arg), D-Arg, taurine (Tau), β-Ala, L/D-β-Phe, α-aminobutyric acid (α-ABA), β-homoserine (β-HSer), and Gly were made in DI water and diluted to working concentrations using a balanced salt solution (BSS) containing 125 mM NaCl, 5.9 mM KCl, 1.2 mM MgCl2, 2.4 mM CaCl2, and 25 mM HEPES at pH 7.4. 2,3-Naphthalenedicarboxaldehyde (NDA, Thermo Fisher Scientific, Waltham, MA) was made at a concentration of 5 mM in a 1:1 (v/v) mixture of ACN and 15 mM borate buffer, pH 9.0. Sodium cyanide (NaCN) was prepared in the same borate buffer to a working concentration of 40 mM. Islet secretions and standards were in BSS and derivatized using 10:0.5:0.5:1 (v/v/v/v) sample: 40 mM NaCN: 24 μM internal standards (IS): 5 mM NDA in a 0.2 mL polypropylene tube. The tube was vortexed for 30 s and placed in the sample tray of the CE instrument where the temperature was held at 25 °C and reactions were allowed to proceed for 30 min prior to injection. As described further in the text, various IS were used to normalize migration times and peak areas of the analytes.
CE-LIF instrumentation operation
MEKC experiments were carried out on a Beckman-Coulter PA800 with a laser-induced fluorescence (LIF) module. A 60 cm (50 cm effective) length of 25 μm ID fused-silica capillary (Polymicro Technologies, Phoenix, AZ) was used for separation. A 100 mW, 450 nm laser diode (Lilly Electronics, Hubei, P. R. China) was used as the excitation source with a 0.6 neutral density filter to reduce the laser intensity to ∼1 mW prior to the capillary. A 480 ± 20 nm bandpass filter and 457 nm notch filter were placed prior to the detector. Data acquisition was at 4 Hz.At the start of each day, the capillary was rinsed with 1 M NaOH, 0.1 M NaOH, and DI water, each for 10 min at 50 psi. Between analyses, the capillary was rinsed with 0.1 M NaOH followed by the running buffer, each for 5 min at 50 psi. Samples were injected by applying 1 psi for 15 s, followed by a wait step in DI water for 6 s. At the end of the day, the capillary was rinsed with 0.1 M NaOH and DI water for 10 min at 50 psi each. The capillary was stored in DI water vials when not in use. For all runs, the cartridge and sample storage temperatures were held at 25 °C.
Isolation and incubation of islets of Langerhans
All animal experiments were performed under guidelines approved by the Florida State University Animal Care and Use Committee, protocol #1813. Islets were obtained by digesting the pancreas from two male CD-1 mice (∼30 g) with collagenase as previously described.12,26 The islets from both mice were combined and incubated at 37 °C with 5% CO2 in RPMI 1640 media containing 11 mM glucose, 10% serum, 100 units per mL penicillin, 100 μg mL−1 streptomycin, and 10 μg mL−1 gentamicin. Media was changed every 48 h. Islets were tested within 5 days of isolation after an initial overnight recovery period.For secretion experiments, 50 islets were removed from the culture media and rinsed twice with prewarmed BSS containing 3 mM glucose. The rinsed islets were then incubated in 100 μL of this solution at 37 °C with 5% CO2. After 1 hour, 90 μL of the supernatant was removed and replaced with 90 μL of 22.2 mM glucose to give a final concentration of 20 mM glucose. The islets were again incubated for 1 hour at 37 °C with 5% CO2 before removing another 90 μL of the BSS. One 5 μL aliquot from each supernatant was derivatized as described in the “Amino acid derivatization” section. For spiking experiments, 0.5 μL of standard D-Ser solution was added to a new 5 μL aliquot from the 90 μL supernatant and derivatized. The final spiked concentrations of D-Ser are given in the text. As stated in the text, in some studies, islets were incubated with the D-amino acid oxidase (DAO) inhibitor, 4H-thieno[3,2-b]pyrrole-5-carboxylic acid (Sigma-Aldrich, St. Louis, MO). The inhibitor was diluted to 500 nM in BSS with 3 mM or 20 mM glucose and 50 islets were incubated in these solutions, at 37 °C and 5% CO2 for two hours prior to analysis of the supernatant. The standards and islet samples were stored at 4 °C prior to derivatization.
Data analysis
Electropherograms were analyzed using 32 Karat™ software (Beckman Coulter, Brea, CA). The software was used to calculate the peak area of specific migration time windows that coincided with each standard. Calibration curves were obtained by plotting the background subtracted peak area of each standard normalized to the background subtracted peak area of the closest IS against its pre-derivatized concentration. Regression equations were calculated by linear least-squares. Limits of detection (LOD) were determined using 3 times the standard deviation (SD) of the background peak area divided by the slope of the calibration curve. Theoretical plates were calculated as previously described.25 The resolution between peaks was calculated using eqn (1). Differences in background subtracted, normalized peak areas were considered significant when p < 0.05 using a one-tailed Student's t-test.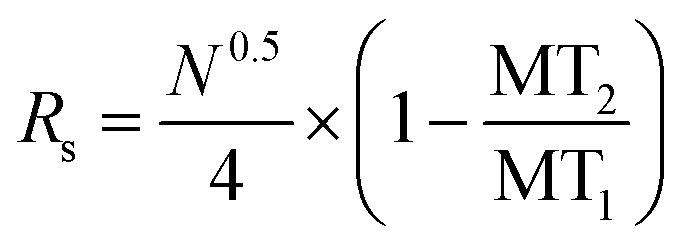 | (1) |
Results and discussion
In previous work, we developed an MEKC method for the resolution of numerous small molecules released from islets12 and then applied this separation method to monitoring small molecule release from islets on a microfluidic system.26 The fluorophore, NDA, was utilized for its fast derivatization of primary amines to isoindole derivative and its subsequent high sensitivity with LIF detection. Since detection of DAA were of interest, a method to introduce chiral resolution to our previous separation was needed. While cyclodextrins have been used to induce chiral separation,15,27–31 our initial attempts at using this additive were not successful. Further, since our goal is to incorporate this chiral separation into a microfluidic system for monitoring release from islets,26 we did not want to utilize nonaqueous separation methods.15 We then turned to other reagents that have been proposed to induce chiral separation, including the use of the bile salt, sodium deoxycholate (SDC) in conjunction with the traditional surfactant used in MEKC, SDS. SDC is known to form mixed micelles with SDS and has been used for chiral MEKC separations.31–33 In this report, an MEKC method containing SDC and SDS was developed and optimized for resolution of several DAA and applied to the investigation of small molecules released from murine islets of Langerhans.Optimization of separation conditions
While our initial goal was to develop a separation method to resolve enantiomeric pairs of most amino acids, D- and L-Ser was chosen as the critical pair due to the known biological role of D-Ser in binding to NMDAR. Also, our previous work12,26 indicated that L-Ser had a relatively small partition coefficient when utilizing SDS and would therefore be more difficult to resolve compared to later-eluting pairs.Initial optimization of the separation parameters included the use of our previously reported phosphate separation buffer.12,26 However, due to the pH being close to the limit of this buffer system, we changed to the use of a boric acid buffer. Initial optimization of the buffer system included testing a range of boric acid, pH, and SDS concentrations. Throughout this optimization, the capillary i.d. was held at 25 μm to limit the effects of Joule heating. The sample used for the optimization of the separation buffer consisted of 19 amines: L-Ser, D-Ser, L-Asn, L-Thr, L-Gln, L-Glu, L-Asp, L-His, Gly, L-Ala, L-Tyr, L-Met, L-Val, GABA, L-Ile, L-Leu, L-Phe, L-Trp, and L-Arg. This sample was used to reduce complications of an initially large number of analytes while still containing the critical pair (D- and L-Ser) as well as the other primary amines that were previously used in our achiral MEKC separations.12,26
Upon completion of these initial experiments, we found that a 150 mM boric acid buffer at pH 9.0 with 45 mM SDS using a 50 cm effective (60 cm total) capillary with a separation voltage of 29 kV was ideal for resolution of the L-amino acids. Once this initial non-chiral separation was optimized, the effect of SDC on the separation was examined. Without SDC, the critical pair was not resolved and the resolution did not increase above 1.0 until after SDC was increased above 25 mM. As shown in Fig. 1A, the effect of increasing SDC concentration (25 to 40 mM from the bottom to top) was a lengthening of the migration times and elution window. As the SDC concentration increased, resolution between the critical pair increased from 1.0 to 1.5 which can be seen in Fig. 1B. However, as the concentration of SDC increased above 35 mM, the resolution of other primary amines of interest decreased, notably the loss of Gly and GABA. Due to the balance between the resolution of the critical pair and the other amines, along with the tradeoff in increased migration times at high SDC concentrations, 35 mM was deemed optimal with the resolution on the critical pair at 1.5.
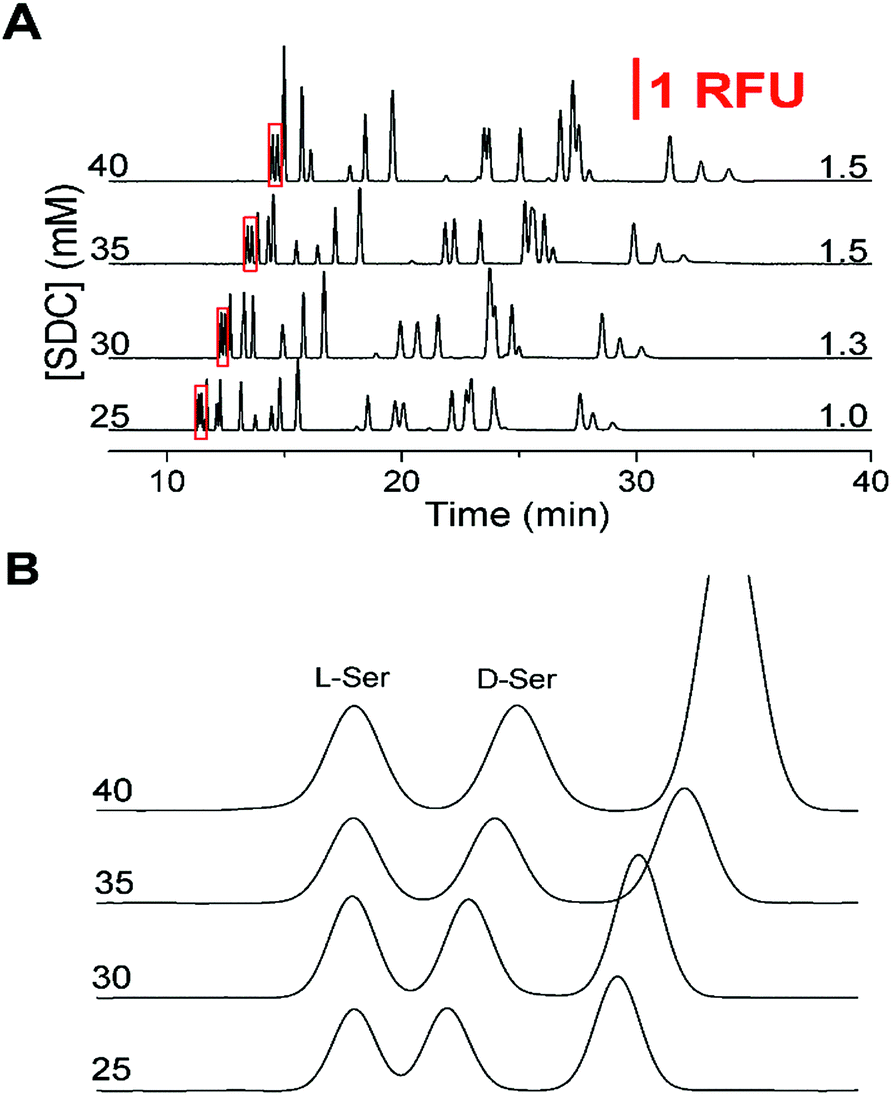 | ||
| Fig. 1 Optimization of separation buffer with corresponding critical pair (D/L-Ser) resolution to the right of the traces. The vertical scale bar showing 1 relative fluorescence unit corresponds to the first part of the figure. All electropherograms were offset for clarity. (A) The SDC concentration is shown to the left of the y-axis for the corresponding electropherograms. The separation buffer was 150 mM boric acid (pH 9.0) with 45 mM SDS. Resolution between the critical pair (D/L-Ser) is shown to the right. (B) Zoomed in view of the critical pair (red boxed regions in (A)) to highlight the resolution. Electropherograms were aligned to the L-Ser peak for ease of viewing. | ||
After determination of the optimized surfactant concentrations, the effects of several other parameters were re-examined. Not shown here, the effects of pH were examined when the separation buffer consisted of 150 mM boric acid, 45 mM SDS, and 35 mM SDC by varying the pH from 8.5 to 9.8. At a pH less than 9.0, poor resolution of the critical pair was observed. With higher buffer pH, the critical pair was well resolved, although at the expense of increased run times. A suitable compromise between resolution of the critical pair and run time was at a separation pH of 9.2. At this pH, the resolution on the critical pair was 1.7 while the resolution on the majority of the other primary amines were satisfactory. Using the optimal pH, the SDS concentration was again examined between 30 to 50 mM; however, there was little further improvement in the resolution of the critical pair.
Following optimization of the separation parameters, a sample containing 36 primary amines containing 13 enantiomeric pairs was tested. A representative separation is shown in Fig. 2A, with zoomed in regions corresponding to each IS region in Fig. 2B–E. With the optimized conditions, 17/36 analytes had a resolution ≥ 1.0 to their nearest neighbour (Table 1). All had plate numbers approaching 300000 with asymmetry values close to 1. With respect to the DAAs, D-Ser and D-Tyr had resolution > 1.5, while Met, Val, and Trp had a resolution between 0.8 and 1.1. No resolution was obtained for the enantiomer pairs of Glu, Asp, Aln, Leu, Phe, and Arg and are listed as not resolved (N.R.) in Table 1. Due to D/L-Glu having variable migration times, although D-Thr is shown in Fig. 2, it was unresolved in other separations and is listed as peak 6* in Table 1. Due to the difficulty in finding conditions that resolved all enantiomeric pairs, and with our goal of resolution of D- and L-Ser satisfied, we then tested the linearity and LOD of the analytes using this separation buffer.
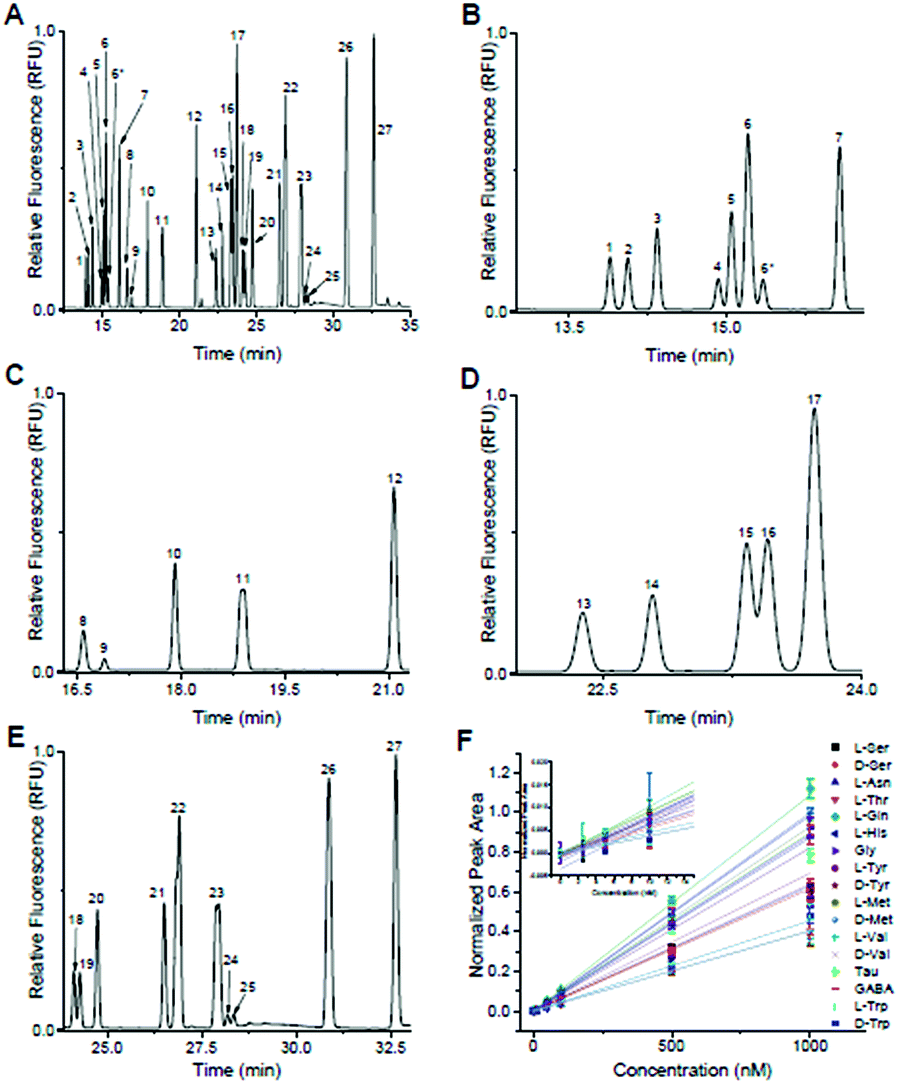 | ||
| Fig. 2 Optimized separation of primary amines. Peak identification: (1) L-Ser; (2) D-Ser; (3) L-Asn; (4) L-Thr; (5) L-Gln; (6) D-Gln, D,L-Glu; (6*) D-Thr; (7) β-HSer (IS); (8) D/L-Asp; (9) L-His; (10) Gly; (11) D/L-Ala; (12) α-ABA (IS); (13) L-Tyr; (14) D-Tyr; (15) L-Met; (16) D-Met; (17) β-Ala (IS); (18) L-Val; (19) D-Val; (20) Tau; (21) GABA; (22) L-Ile, D/L-Leu; (23) D/L-Phe; (24) L-Trp; (25) D-Trp; (26) D/L-β-Phe (IS); (27) D/L-Arg. (A) Full electropherogram of 36 primary amines utilizing the optimized separation parameters of 150 mM boric acid (pH 9.2), 45 mM SDS, and 35 mM SDC using 29 kV across a 60 cm (total length) capillary with i.d. of 25 μm. (B) Zoomed in region of the electropherogram from (A) containing “early” migrating primary amines, L-Ser to D-Thr. Due to D/L-Glu having variable MT, D-Thr did not resolve in later separations. These primary amines were normalized to β-HSer (peak 7). (C) Zoomed in region from (A) containing “mid-early” migrating primary amines, L-Asp to D-Ala. These peaks were normalized to α-ABA (peak 12). (D) Zoomed in region from (A) containing “mid-late” migrating primary amines, L-Tyr to D-Met. These peaks were normalized to β-Ala (peak 17). (E) Zoomed in region from (A) containing “late” migrating species, L-Val to D-Arg. These peaks were normalized to D/L-β-Phe (peak 26). (F) Calibration curves for the 17 primary amines that did not co-migrate with any other peaks. | ||
| Peak ID | Peakc | LOD (nM) | N (×105) | R s | Normalized MT % RSDc | Normalized peak area % RSDd |
|---|---|---|---|---|---|---|
| a Plate numbers (N) calculated25 for analytes at 1000 nM. b Resolution (Rs) calculated using eqn (1) for analytes at 1000 nM. c The % RSD of the migration times (MT) normalized to the four IS across all calibration concentrations. Values shown for the IS are the raw % RSD values. d The % RSD of the normalized peak areas for the primary amines using 500 nM standards. Values shown for the IS are the raw % RSD values. | ||||||
| L-Ser | 1 | 4 | 3.5 | 1.7 | 0.37 | 4.9 |
| D-Ser | 2 | 4 | 3.4 | 1.7 | 0.38 | 2.7 |
| L-Asn | 3 | 1 | 3.7 | 2.9 | 0.45 | 2.9 |
| L-Thr | 4 | 3 | 3.5 | 1.2 | 0.16 | 3.4 |
| L-Gln | 5 | 2 | 3.5 | 1.2 | 0.17 | 3.2 |
| D-Gln | 6 | N.R. | ||||
| D/L-Glu | 6 | N.R. | ||||
| D-Thr | 6* | N.R. | ||||
| β-HSer | 7 | IS | 3.7 | 5.3 | 1.6 | 4.8 |
| D/L-Asp | 8 | N.R. | ||||
| L-His | 9 | 0.4 | 2.8 | 1.5 | 0.41 | 7.0 |
| Gly | 10 | 8 | 3.5 | 7.3 | 0.30 | 4.6 |
| D/L-Ala | 11 | N.R. | ||||
| α-ABA | 12 | IS | 3.6 | 8.4 | 2.1 | 4.3 |
| L-Tyr | 13 | 2 | 4.0 | 2.7 | 0.20 | 4.6 |
| D-Tyr | 14 | 3 | 4.2 | 2.8 | 0.12 | 3.5 |
| L-Met | 15 | 0.7 | 5.1 | 0.8 | 0.08 | 3.3 |
| D-Met | 16 | 3 | 5.0 | 0.8 | 0.07 | 2.3 |
| β-Ala | 17 | IS | 4.4 | 1.8 | 2.4 | 5.0 |
| L-Val | 18 | 2 | 4.6 | 1.1 | 0.59 | 9.9 |
| D-Val | 19 | 3 | 2.5 | 2.0 | 0.56 | 11.1 |
| Tau | 20 | 3 | 4.3 | 2.9 | 0.49 | 8.7 |
| GABA | 21 | 1 | 4.7 | 2.1 | 0.36 | 7.9 |
| L-Ile | 22 | N.R. | ||||
| D/L-Leu | 22 | N.R. | ||||
| D/L-Phe | 23 | N.R. | ||||
| L-Trp | 24 | 0.3 | 4.7 | 0.9 | 0.27 | 3.5 |
| D-Trp | 25 | 0.6 | 5.4 | 1.0 | 0.23 | 2.9 |
| D/L-β-Phe | 26 | IS | 3.4 | 7.6 | 3.0 | 10.6 |
| D/L-Arg | 27 | N.R. | ||||
Included in the 36-component sample shown in Fig. 2 were L-β-HSer, α-ABA, L-β-Ala, and D,L-β-Phe (did not resolve), which were used as IS. We used these analytes as IS because, to the best of our knowledge, they are not known to be released from islets. L-β-HSer was used as the IS for analytes that eluted “early” (D,L-Ser, L-Asn, D,L-Thr, D,L-Gln, D,L-Glu, Fig. 2B); α-ABA for analytes that eluted “mid-early” (D,L-Asp, L-His, Gly, D,L-Ala, Fig. 2C); L-β-Ala for analytes that eluted “mid-late” (D,L-Tyr, D,L-Met, Fig. 2D); and D,L-β-Phe for analytes that eluted “late” (D,L-Val, Tau, GABA, L-Ile, D,L-Leu, D,L-Phe, D,L-trp, and D,L-Arg, Fig. 2E). Calibration curves were obtained for those analytes that did not co-migrate with other standards. These included D-Ser, L-Ser, L-Asn, L-Thr, L-Gln, L-His, Gly, L-Tyr, D-Tyr, L-Met, D-Met, L-Val, D-Val, Tau, GABA, L-Trp, and D-Trp. Calibrations were obtained by derivatizing these analytes at concentrations of 1000, 500, 100, 50, 10, 5, 2.5, and 0 nM, while maintaining the concentrations of the IS at 1 μM; the derivatization and analysis of each concentration value was performed in triplicate. The normalized peak area of each analyte to its IS vs. concentration of analyte were linear (r2 > 0.99 by linear regression) for all analytes over this concentration range (Fig. 2F) and limits of detection ranged from 0.3–8 nM (Table 1).
High intra- and inter-day reproducibility were observed for migration times normalized to their respective IS with relative standard deviations (RSD) < 1% and IS migration time % RSD < 5% as seen in Table 1. Similarly, normalized peak areas were reproducible with the majority of the RSD < 5% although most increased at the lower concentrations tested. One difficulty in obtaining higher reproducibility were differences in the NDA reaction times between samples. These differences led to differences in peak areas and contributed to the higher RSD especially at low analyte concentrations. Nevertheless, upon completion of the calibration curves, this method was used to examine glucose-stimulated release of amino acids from murine islets of Langerhans.
Determination of amino acids secreted from islets of Langerhans
With the separation buffer optimized, the release of DAAs from islets was examined by derivatizing the extracellular solution of two sets of 50 murine islets of Langerhans after incubation in 3 mM glucose for 1 hour and 20 mM glucose for 1 hour. The electropherograms from one of these samples are shown in Fig. 3A and B with the second set shown in the ESI, Fig. S1.† The levels from both of these trials were quantified using the calibration curves in Fig. 2F and the values are shown in Table 2. Those peaks that were not above a detectable concentration are labelled as “N.D.”, while those that were not resolved in the calibration curves are again labelled N.R. In general, the trends of the released components using this new method agreed with our previously published reports:12,26 all resolved and detectable analytes decreased with increasing glucose concentration except L-Gln which previously was observed to increase in the elevated glucose levels.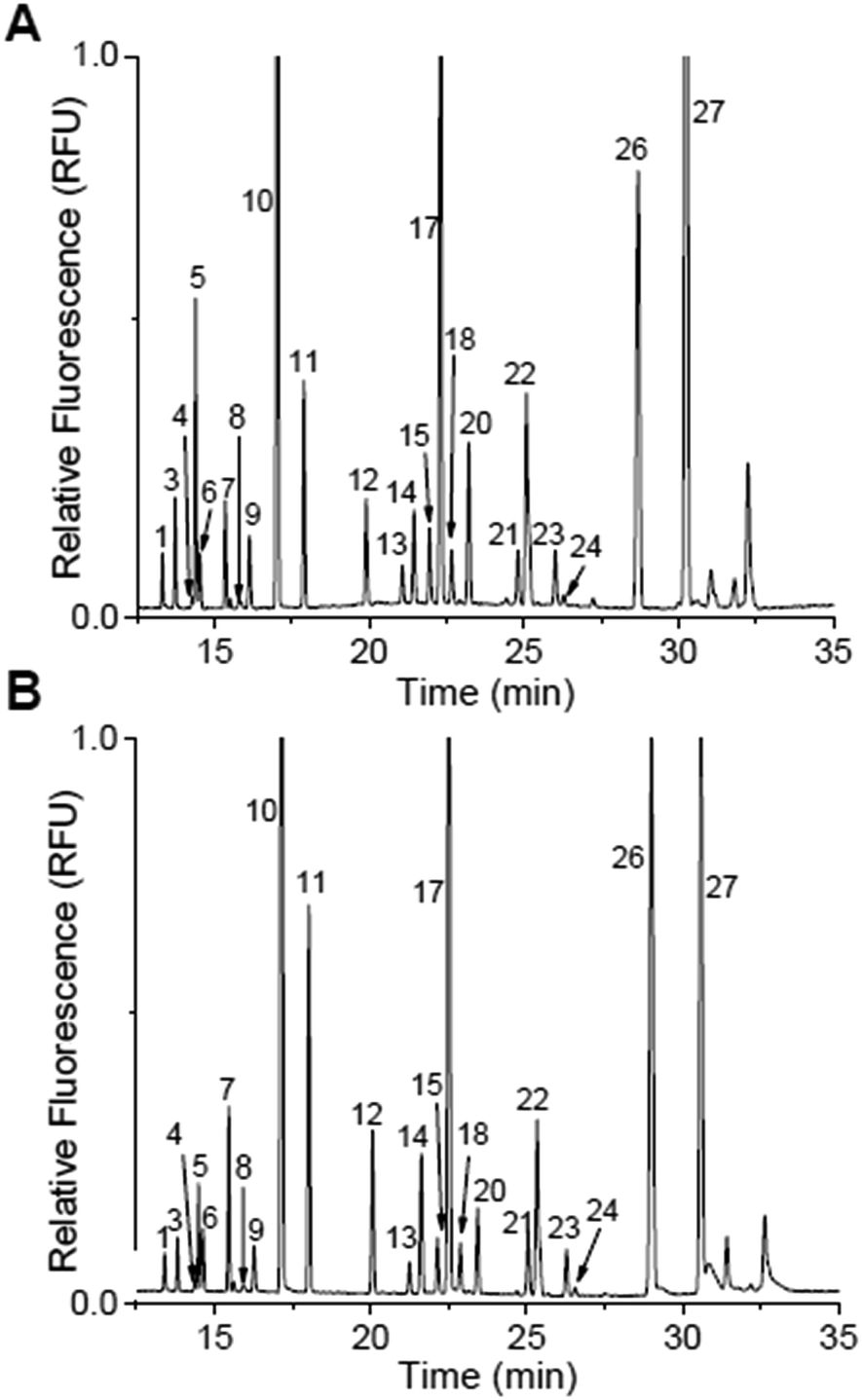 | ||
| Fig. 3 Islet secretions stimulated by glucose. Peak identification is the same in both electropherograms as in Table 1 and Fig. 2. (A) Representative electropherograms of islet secretions from 50 murine islets that were stimulated with 3 mM glucose for 1 h. (B) The same 50 islets from (A) were then incubated in 20 mM glucose for 1 h. | ||
| Peak ID | Peak number | Trial 1 | Trial 2 | ||
|---|---|---|---|---|---|
| 3 mM | 20 mM | 3 mM | 20 mM | ||
| L-Ser | 1 | 739 (8) | 298 (10) | 1336 (12) | 508 (17) |
| D-Ser | 2 | N.D. | |||
| L-Asn | 3 | 1098 (22) | 289 (7) | 1949 (39) | 465 (16) |
| L-Thr | 4 | 259 (9) | 142 (10) | 370 (20) | 173 (16) |
| L-Gln | 5 | 2463 (82) | 481 (4) | 4996 (126) | 900 (16) |
| D-Gln | 6 | N.R. | |||
| D/L-Glu | 6 | N.R. | |||
| D-Thr | 6 | N.R. | |||
| β-HSer | 7 | IS | |||
| D/L-Asp | 8 | N.R. | |||
| L-His | 9 | 1076 (144) | 439 (33) | 2136 (351) | 764 (84) |
| Gly | 10 | 13925 (966) |
7950 (1116) | 25394 (2118) |
13013 (1579) |
| D/L-Ala | 11 | N.R. | |||
| α-ABA | 12 | IS | |||
| L-Tyr | 13 | 76 (2) | 57 (4) | 86 (4) | 57 (2) |
| D-Tyr | 14 | IS | |||
| L-Met | 15 | 84 (2) | 57 (3) | 90 (4) | 48 (1) |
| D-Met | 16 | N.D. | |||
| β-Ala | 17 | IS | |||
| L-Val | 18 | 235 (28) | 181 (13) | 343 (6) | 206 (7) |
| D-Val | 19 | N.D. | |||
| Tau | 20 | 306 (14) | 124 (1) | 569 (13) | 321 (11) |
| GABA | 21 | 92 (4) | 86 (2) | 231 (7) | 188 (6) |
| L-Ile | 22 | N.R. | |||
| D/L-Leu | 22 | N.R. | |||
| D/L-Phe | 23 | N.R. | |||
| L-Trp | 24 | 35 (4) | 21 (2) | 49 (2) | 20 (2) |
| D-Trp | 25 | N.D. | |||
| D/L-β-Phe | 26 | IS | |||
| D/L-Arg | 27 | N.R. | |||
As for the DAAs (excluding D-Ser), none were observed at a quantifiable level for the 3- or 20 mM glucose sample. It should be noted that when the samples shown in Fig. 3 and S1† were separated, D-Tyr (peak 14) was also included as an extra IS. The reason for using this DAA as an IS was that it had not been reported to be found in other tissues.34,35
In the 20 mM glucose sample from both islet samples, there was a small peak that migrated at a similar time as the D-Ser standard. Spiking experiments were performed to test the identity of this peak (Fig. 4). Different aliquots of these samples were spiked with D-Ser to final concentrations of 0 to 30 nM. The 0 nM D-Ser spike was used to account for any dilution resulting from the spike. As seen in Fig. 4, in both islet samples, the small peak co-migrated with the spiked standards. Although we cannot definitively state that this peak was D-Ser, the co-migration of the peak with the standards is evidence towards its presence. The samples from both sets of islets incubated at 3 mM glucose did not have as clear of a peak as the 20 mM sample.
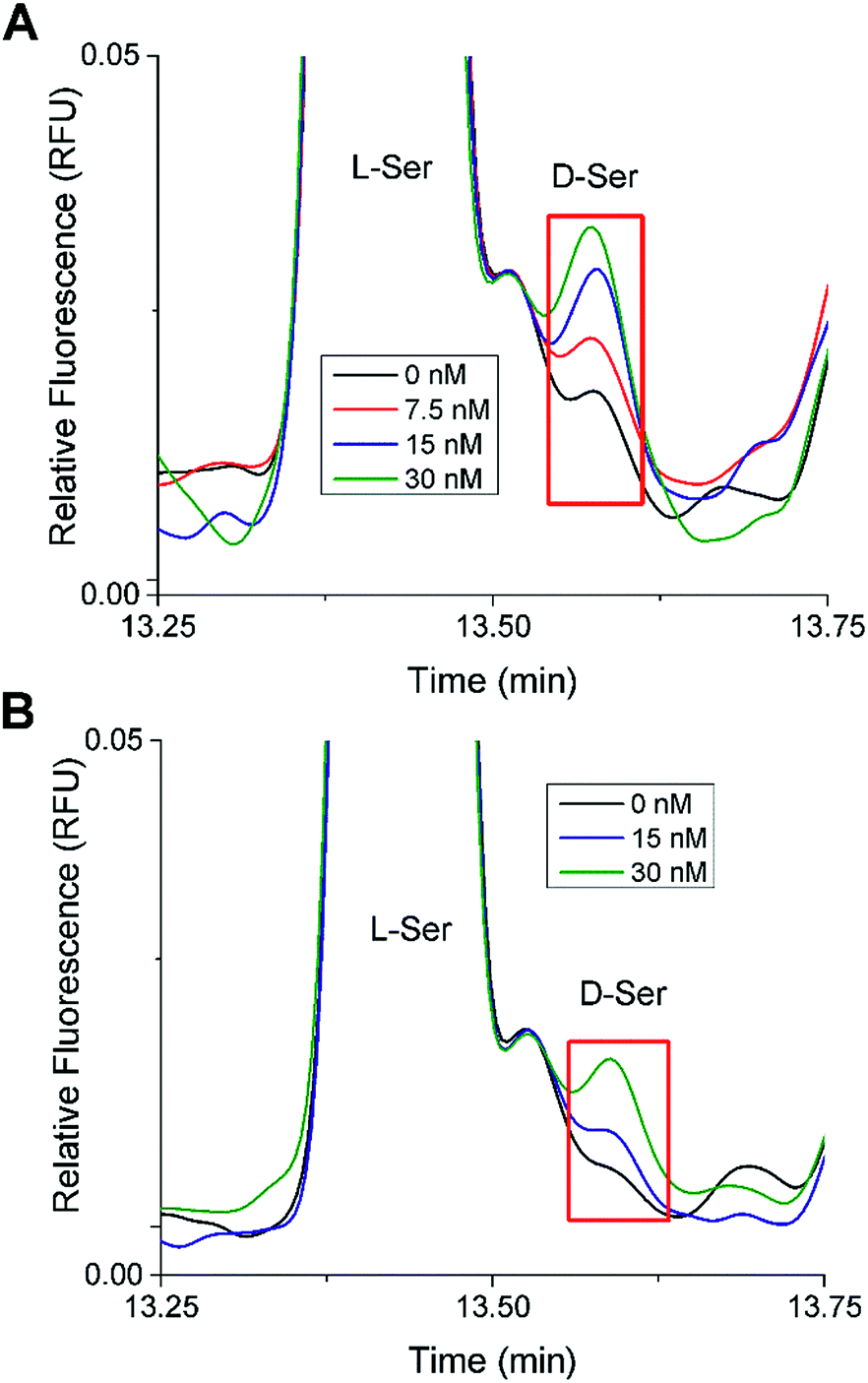 | ||
| Fig. 4 D-Ser spiking experiments. Shown are stacked electropherograms from spiking experiments where aliquots of the islet samples incubated in 20 mM glucose were spiked with D-Ser standard to produce final D-Ser concentrations from 0 to 30 nM as shown. The electropherograms were aligned to L-Ser for comparison. (A) The electropherogram from Fig. 3B (Trial 1). (B) The electropherogram from Fig. S1B† (Trial 2). | ||
In a further attempt to increase the amount of DAAs released from islets, different islet samples were incubated with an inhibitor of DAO, with the expectation that it should inhibit the intracellular enzyme responsible for oxidizing most DAA, and thereby result in increased amounts of DAAs released.36 Unfortunately, no significant changes in the electropherograms compared to those shown in Fig. 3 were observed. This result is difficult to interpret because to the best of our knowledge, DAO has not been reported directly in islets, although increased levels of D-Ser and D-Ala were found in the whole pancreas of DAO knockout mice.16
Conclusions
The method presented in this article was able to resolve the enantiomers of Ser while preserving resolution for numerous small molecules, such as GABA, that may play a role in islet communication. The method was also able to resolve other DAAs which may be of future interest for cell secretion studies. While the analysis of islet secretions at high glucose seemed to point to the presence of D-Ser in low nanomolar concentrations, multiple unknown peaks in the general migration window hinders our ability to confidently identify it. As for DAA such as D-Asp, D-Glu and D-Ala, due to the co-migration of those peaks with their enantiomeric counterpart, we cannot definitively state if any of these known agonists for NMDARs were released utilizing our current method. However, this separation method is suitable for resolving D-Ser at low nanomolar concentrations which is significant due to its affinity for the Gly binding site in NMDARs, and with previously reported24 measurements of Srr in islets. It should be noted that we observed very high levels of Gly released from islets at both 3 and 20 mM glucose. Further optimization is required with both the separation method and sampling procedure to increase the resolution between D-Ser and the other unidentified primary amines to aid in quantitation.Conflicts of interest
The authors declare that they have no conflicts of interest to declare.Acknowledgements
MGR gratefully acknowledges support from the Pfeiffer Professorship fund from Florida State University. Research reported in this publication was supported by the National Institute of Diabetes of the National Institutes of Health and Digestive and Kidney Diseases under Award Number R01DK080714 and the National Institute On Drug Abuse of the National Institutes of Health under Award Number R21DA044442. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors would like to thank I-An Wei for the initial work on the use of multiple IS in the separation.Notes and references
- World Health Organization, Global Report on Diabetes, ISBN: 978 92 4 156525 7, 2016 Search PubMed.
- J. Girard, Biochimie, 2017, 143, 33–36 CrossRef CAS PubMed.
- R. H. Unger, R. E. Dubbs and L. Orci, Annu. Rev. Physiol., 1978, 40, 307–343 CrossRef CAS PubMed.
- L. S. Satin and T. A. Kinard, Endocrine, 1998, 8, 213–223 CrossRef CAS PubMed.
- E. Gylfe and A. Tengholm, Diabetes, Obes. Metab., 2014, 16, 102–110 CrossRef CAS PubMed.
- R. Rodriguez-Diaz, D. Menegaz and A. Caicedo, J. Physiol., 2014, 16, 3413–3417 CrossRef PubMed.
- E. Gylfe, E. Grapengiesser, H. Dansk and B. Hellman, Pancreas, 2012, 41, 258–263 CrossRef CAS PubMed.
- R. Yan-Do, E. Duong, J. E. M. Fox, X. Q. Dai, K. Suzuki, S. Khan, A. Bautista, M. Ferdaoussi, J. Lyon, X. C. Wu, S. Cheley, P. E. MacDonald and M. Braun, Diabetes, 2016, 65, 2311–2321 CrossRef CAS PubMed.
- C. Li, C. Liu, I. Nissim, J. Chen, P. Chen, N. Doliba, T. Zhang, I. Nissim, Y. Daikhin, D. Stokes, M. Yudkoff, M. J. Bennett, C. A. Stanley, F. M. Matschinsky and A. Naji, J. Biol. Chem., 2013, 288, 3938–3951 CrossRef CAS PubMed.
- A. Caicedo, Semin. Cell Dev. Biol., 2013, 24, 11–21 CrossRef CAS PubMed.
- P. Rorsman, P. O. Berygren, K. Bokvist, H. Ericson, H. Möhler, C. G. Östenson and P. A. Smith, Nature, 1989, 341, 233–236 CrossRef CAS PubMed.
- X. Wang, L. Yi, C. Guillo and M. G. Roper, Electrophoresis, 2015, 36, 1172–1178 CrossRef CAS PubMed.
- Y. Kera, H. Aoyama, H. Matsumura, A. Hasegawa, H. Nagasaki and R. Yamada, Biochim. Biophys. Acta, 1995, 1243, 282–286 CrossRef.
- M. Hiasa and Y. Moriyama, Biol. Pharm. Bull., 2006, 29, 1251–1253 CrossRef CAS.
- N. Ota, S. S. Rubakhin and J. V. Sweedler, Biochem. Biophys. Res. Commun., 2014, 447, 328–333 CrossRef CAS PubMed.
- Y. Miyoshi, K. Hamase, Y. Tojo, M. Mita, R. Konno and K. Zaitsu, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2009, 877, 2506–2512 CrossRef CAS PubMed.
- S. Karakawa, K. Shimbo, N. Yamada, T. Mizukoshi, H. Miyano, M. Mita, W. Lindner and K. Hamase, J. Pharm. Biomed. Anal., 2015, 115, 123–129 CrossRef CAS PubMed.
- M. Horio, M. Kohno, Y. Fujita, T. Ishima, R. Inoue, H. Mori and K. Hashimoto, Neurochem. Int., 2011, 59, 853–859 CrossRef CAS PubMed.
- J. P. Mothet, A. T. Parent, H. Wolosker, R. O. Brady Jr, D. J. Linden, C. D. Ferris, M. A. Rogawski and S. H. Snyder, Proc. Natl. Acad. Sci., 2000, 97, 4926–4931 CrossRef CAS.
- L. V. Kalia, S. K. Kalia and M. W. Salter, Lancet Neurol., 2008, 7, 742–755 CrossRef CAS PubMed.
- J. Marquard, S. Otter, A. Welters, A. Stirban, A. Fischer, J. Eglinger, D. Herebian, O. Kletke, M. S. Klemen, A. Stožer, S. Wnendt, L. Piemonti, M. Köhler, J. Ferrer, B. Thorens, F. Schliess, M. S. Rupnik, T. Heise, P. O. Berggren, N. Klocker, T. Meissner, E. Mayatepek, D. Eberhard, M. Kragl and E. Lammert, Neonat. Med., 2015, 21, 363–372 CAS.
- T. Gonoi, N. Mizunol, N. Inagakis, H. Kuromil, Y. Seinol, J. Miyazakill and S. Seino, J. Biol. Chem., 1994, 269, 16989–16992 CAS.
- E. Molnár, A. Váradi, R. A. McIlhinney and S. J. H. Ashcroft, FEBS Lett., 1995, 371, 253–257 CrossRef.
- A. D. Lockridge, D. C. Baumann, B. Akhaphong, A. Abrenica, R. F. Miller and E. U. Alejandro, Islets, 2016, 8, 195–206 CrossRef CAS PubMed.
- J. P. Foley and J. G. Dorsey, Anal. Chem., 1983, 55, 730–737 CrossRef CAS.
- X. Wang, L. Yi and M. G. Roper, Anal. Chem., 2016, 88, 3369–3375 CrossRef CAS PubMed.
- D. Rosenberg, E. Kartvelishvily, M. Shleper, C. M. C. Klinker, M. T. Bowser and H. Wolosker, FASEB J., 2010, 24, 2951–2961 CrossRef CAS PubMed.
- H. Miao, S. S. Rubakhin and J. V. Sweedler, Anal. Chem., 2005, 77, 7190–7194 CrossRef CAS PubMed.
- C. M. Ciriacks and M. T. Bowser, Anal. Chem., 2004, 76, 6582–6587 CrossRef CAS PubMed.
- K. DeSilva and T. Kuwana, Biomed. Chromatogr., 1997, 11, 230–235 CrossRef CAS PubMed.
- D. L. Kirschner and T. K. Green, J. Sep. Sci., 2009, 32, 2305–2318 CrossRef CAS PubMed.
- M. G. Khaledi, J. G. Bumgarner and M. Hadjmohammadi, J. Chromatogr. A, 1998, 802, 35–47 CrossRef CAS PubMed.
- S. Zhao, Y. Song and Y.-M. Liu, Talanta, 2005, 67, 212–216 CrossRef CAS PubMed.
- M. Yamanaka, Y. Miyoshi, H. Ohide, K. Hamase and R. Konno, Amino Acids, 2012, 43, 1811–1821 CrossRef CAS PubMed.
- Y. Kiriyama and H. Nochi, Scientifica, 2016, 2016, 1–9 CrossRef PubMed.
- S. M. Smith, J. M. Uslaner, L. Yao, C. M. Mullins, N. O. Surles, S. L. Huszar, C. H. McNaughton, D. M. Pascarella, M. Kandebo, T. M. Hinchliffe, T. Sparey, N. J. Brandon, B. Jones, S. Venkatraman, M. B. Young, N. Sachs, M. A. Jacobson and P. H. Hutson, J. Pharmacol. Exp. Ther., 2009, 328, 921–930 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ay02471e |
| This journal is © The Royal Society of Chemistry 2019 |