
A simple and effective strategy for detecting artemisinin based on oxidative cyclization of vitamin B1 eliciting fluorescence turn-on†
Wu
Zou
a,
Fuchun
Gong
 *a,
Zhong
Cao
*ab,
Jiaoyun
Xia
a,
Tingting
Gu
a and
Renxing
Deng
a
*a,
Zhong
Cao
*ab,
Jiaoyun
Xia
a,
Tingting
Gu
a and
Renxing
Deng
a
aCollege of Chemistry and Biologic Engineering, Changsha University of Science and Technology, Changsha, 410114, P. R. China. E-mail: gongfc139@163.com; Fax: +86-731-85258733; Tel: +86-731-85258733
bHunan Provincial Key Laboratory of Materials Protection for Electric Power and Transportation, Changsha University of Science and Technology, Changsha 410114, P. R. China
First published on 16th November 2018
Abstract
Artemisinin (ART) and its derivatives are first-line treatment drugs for falciparum malaria and save millions of lives every year. It is very difficult to design specific fluorescent probes for ART because ART has no groups for binding to except for the peroxide group. We find that alkaline-hydrolyzed ART (a-ART) can specifically recognize and react with vitamin B1 (VB1) and produce fluorimetrically detectable thiochromes. Notably, this chemistry is affected by very low concentrations of a-ART in a Tris–HCl buffer solution (pH 7.5) at room temperature, leading to >260-fold enhancement in the blue emission at 442 nm. By making use of the new signaling mechanism, an effective strategy for detecting ART was developed. The fluorescence intensity of the VB1-based probes linearly increased with increasing ART concentration ranging from 1 to 230 μM mL−1, and a detection limit as low as 11.5 nM mL−1 was achieved, making this method more sensitive than other reported UV-vis absorption and electrochemical methods. Moreover, this assay shows good selectivity over other ions and biomolecules. The proposed method was applied for the determination of ART in tablets and dried leaf samples of Artemisia annua L. with satisfactory results, which confirms its great potential for real sample analysis. The designed assay avoids the preparation of fluorescent probes, and all the detection processes are accomplished within 20 min. Therefore, VB1 can offer a simple, highly sensitive and selective sensing model for fluorescence detection of ART.
1. Introduction
Malaria remains a major public health problem in tropical and subtropical countries. In 2015 alone, there were 214 million cases of malaria and an estimated 438![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths worldwide.1 Although the bothersome trend of the resistance to antimalarial drugs is increasing at a phenomenal rate, malaria infections are curable by using different antimalarial drugs. A popular type of antimalarial drug which has successfully drawn great attention from the medical fraternity all these years is artemisinin (ART) and its derivatives.2 As a sesquiterpene lactone compound containing the specific endoperoxide bridge, ART was first discovered in the Chinese medicinal herb Artemisia annua L. and has been successfully used in the treatment of the growing resistance of plasmodium falciparum for more than two decades.3 Currently, ART and its derivatives are still the most important antimalarial drugs available and ART-based combination therapy has been recommended worldwide as first-line treatment for falciparum malaria since 2001.4
000 deaths worldwide.1 Although the bothersome trend of the resistance to antimalarial drugs is increasing at a phenomenal rate, malaria infections are curable by using different antimalarial drugs. A popular type of antimalarial drug which has successfully drawn great attention from the medical fraternity all these years is artemisinin (ART) and its derivatives.2 As a sesquiterpene lactone compound containing the specific endoperoxide bridge, ART was first discovered in the Chinese medicinal herb Artemisia annua L. and has been successfully used in the treatment of the growing resistance of plasmodium falciparum for more than two decades.3 Currently, ART and its derivatives are still the most important antimalarial drugs available and ART-based combination therapy has been recommended worldwide as first-line treatment for falciparum malaria since 2001.4
Quality control and efficacy evaluation are important links to ensure the use of ART in clinic and therefore worthy of use in sciatic investigation. A significant similarity was found in the case of side effects from ART-based medications with those of malaria viz., vomiting, nausea, anorexia and dizziness.5,6 In addition, the growing application of ART and its derivatives in clinic has drawn the interest of analysts in developing sensitive, selective, and simple methods for their determination. Various instrumental methods including liquid chromatography-mass spectrometry (LCMS),7,8 high-performance liquid chromatography (HPLC),9,10 HPLC-evaporative light scattering detection (HPLC-ELSD),11 gas chromatography-flame ionization detection (GC-FID),12 HPLC-peroxyoxalate chemiluminescence (HPLC-POCL),13 HPLC-chemiluminescence (HPLC-CL),14 tandem mass spectrometry detection (HPLC-MS/MS)15 and rapid resolution liquid chromatography triple quadrupole mass spectrometry (RRLC-QQQ)16 are reported for ART determination. However, most of them require sophisticated instrumentation and are time consuming. Additionally, the sensitivities of these methods were not enough to meet the increasing analytical requirements nowadays.
The absence of appropriate chromospheres for UV absorbance or fluorescence makes ART determination very difficult. Fortunately, an endoperoxide moiety in ART allows the electrochemical reduction of the peroxide function, which has become the basis of electrochemical methods.17 For example, a graphene modified electrode combined with polymerized molecularly imprinted membranes was used to detect ART.18 A new electrochemical method, which is based on the selective oxidation of p-aminophenylboronic acid by ART to generate p-aminophenol and subsequent detection of the as-generated p-aminophenol, has also been developed.19 Additionally, high-performance liquid chromatography coupled with reductive electrochemical detection was employed to detect artesunic acid and ART in blood.20
Recently, the introduction of some nanomaterials in electrochemical methods for ART determination made them relatively more sensitive. Several nanostructures such as nanoAu-chitosan,21 PHA/Au NP/HRP/ITO hybrid nanocomposites,22 polyhydroxyalkanoate–gold nanoparticle composites23 and multi-wall carbon nanotubes24 were used to construct electrochemical sensing platforms for ART determination. Additionally, an indirect UV absorption method has been developed and used to detect ART in Chinese herb artemisia.25 Although the sensitivity of these nanostructure-based methods for ART detection is greatly enhanced compared with conventional electrochemical methods, the improvement of the selectivity towards the analytes in complex biological samples remains an enormous challenge for practical application.
Fluorescent probes for the recognition of ions and bioactive molecules have attracted much interest for important and diverse ecological, biological, and clinical applications.26–28 They have the advantages of real-time monitoring with fast response times, high sensitivity, and ease of handling compared to electrochemical and other optical methods. The production of probes usually involves complicated molecular design and tedious synthesis.29,30
Herein, we report a VB1-based fluorogenic probe for the detection of trace amounts of ART in leaf samples from Artemisia annua L. and some commercial products. The proposed method is based on the product determination of VB1 oxidized by ART. Opening of the lactones in ART by alkaline hydrolysis releases the carboxyl group and hydroxyl group, which enable it to bind to VB1 by hydrogen bonding and electrostatic pull, which are beneficial for the improvement of the selectivity of ART to VB1. The newly developed method was validated by performing certified reference material analyses and recovery tests. To the best of our knowledge, this is the first time that VB1 has been applied to ART analysis in leaf samples from Artemisia annua L. with such simple and short sample preparation and analysis.
2. Experimental
2.1 Materials and apparatus
Artemisinin (ART) was purchased from Hunan SWS Company, and vitamin B1 was obtained from Beijing Ouhe Technology Company. ART tablets were bought from Guangzhou Ruibai Pharmaceutical Company (Guangzhou, China). All other chemicals were purchased from Aladdin Chemistry Co. Ltd. (Shanghai, China) and were of analytical grade and were used as received. Double distilled water was employed for all the experiments. The stock solution of ART (100 mM) was prepared in ethanol and the stock solution of vitamin B1 (150 mM) was prepared in Tris–HCl buffer solution (pH 7.5). All the experiments were performed at room temperature.All fluorescence measurements were recorded with a Hitachi F-7000 fluorescence spectrophotometer (Tokyo, Japan) with the excitation slit set at 5 nm band pass and emission at 2.5 nm band pass in 1 cm × 1 cm quartz cells. UV-vis spectra were recorded on a Shimadzu UV-1750 spectrophotometer (Tokyo, Japan). Fourier transform infrared (FTIR) spectra were recorded on a Shimadzu IR Prestige-21 spectrometer (Tokyo, Japan). High performance liquid chromatography coupled with ultraviolet spectrophotometry was used to determine the reaction products (Shanghai, China). Mass spectra of ART and the reaction products of a-ART with VB1 were recorded with a SPECTRO MS (Spike, Germany). A Fangzhong pH S-3C digital pH meter (Chengdu, China) was used to measure pH values of the aqueous solutions and a QL-901 vortex mixer (Haimen, China) was employed to blend the solution. The thermostatic water bath (DF-101s) was purchased from Gongyi Experimental Instruments Factory (Gongyi, China).
2.2 Procedures for detecting ART
A typical ART detection procedure including the hydrolysis of ART, the reaction of a-ART with VB1 and the product determination of VB1 is as follows. Firstly, 0.5 mL NaOH (0.05 M) solution was added dropwise into 2.0 mL ethanol–water systems containing different concentrations of ART and reacted for 5 min. Secondly, 1.0 mL of 2.0 mM VB1 in Tris–HCl buffer was added to the hydrolysate and reacted for an additional 5 min. Subsequently, the reaction solution was mixed with 6.5 mL of pH 7.5 Tris–HCl buffer solution, and the total volume used for measurements is 10 mL. Finally, the fluorescence spectra were collected under excitation at 265 nm. All measurements were carried out at room temperature.2.3 Product analysis
The products in the reaction of ART with VB1 were analyzed on an Agilent-1206 high performance liquid chromatograph equipped with a reverse phase column (WAKO Sil-II5C18, 4.6–200 mm) and a VWD UV-vis detector (eluent, MeCN/H2O = 9/1; flow rate, 10.5 mL min−1; monitored at 260 nm) and on a TSQ Altis mass spectrometer.2.4 Calibration and method validation
A stock solution of 10 mM ART was prepared by dissolving 0.283 g solid ART into 10 mL ethanol–water system (1:1, v/v). ART standard solutions with concentrations ranging from 1.0 μM to 230.0 μM were prepared by diluting the stock solution with the ethanol–water system after which the standard solutions were analyzed as described above. A calibration curve was established by plotting the peak intensities of standard solutions against ART concentrations. To evaluate method precision and accuracy, ART samples were analyzed by spiking ART at three different stated concentrations, 2.5 mM, 5 mM, and 10 mM (n = 6).
2.5 Sample preparation
An appropriate amount of dried leaf of A. annua L. (1.000 g, obtained from Yongzhou Xincheng Biotechnolgy Co. Ltd, Yongzhou, China) was thoroughly ground using an agate mortar. The resulting powder was subjected to ultrasonic vibration for 10 min and then extracted by refluxing for 6 h at 60 °C with 50 mL ethanol two times. After cooling to room temperature, the extract was filtered and washed three times with 20 mL ethanol. Subsequently, the filtrate was combined and dried by evaporation and the residue was dissolved in 25 mL ethanol and used as a stock solution. This solution was analyzed directly by UV-vis absorption and fluorescence measurements after it was diluted 20 times with the ethanol–water system.Twenty tablets containing naphthoquine phosphate and ART were randomly chosen and accurately weighed singly to measure the average weight of each tablet. The procedure for analysis of the content of ART in the tablets is as follows: firstly, the whole tablets were pulverised into fine powder using an agate mortar. Secondly, an appropriate amount of the powder (100 mg) was accurately measured into a 50 mL volumetric flask, 15 mL of absolute ethanol was added and the mixture sonicated for 10 min. The resulting mixture in the flask was shaken vigorously as different amounts of ethanol were added to extract the drugs. The solution in the flask was made up to the mark with absolute ethanol. The resulting mixture was then filtered using Whatman filter paper No. 45. The first 10 mL portion of the filtrate was discarded. The resulting drug extract solution with a concentration of 20 mg mL−1 was diluted stepwise to obtain a working concentration (200 μg mL−1) and an appropriate amount of aliquot was subjected to analysis as described in the general procedure above.
2.6 Selectivity investigation
The selectivity of this assay for ART was evaluated by determining the fluorogenic response of VB1 to some ions (Fe3+, Cu2+, Co2+, Zn2+, NO3− and PO43−) and biomolecules (glucose, Lys, Cys, VC, VB6, H2O2 and chlorophyll) both in the absence and presence of ART. In detail, 15 μL of 1 mM EDTA, 15 μL of 0.1 mM a-ART, 15 μL of various concentrations of these ions and biomolecules and 95 μL of 1 mM VB1 were added sequentially into 850 μL Tris–HCl buffer (pH 7.5). The emission spectra were recorded after reaction at 25 °C for 5 min. For the detection of the fluorescence changes of these possible interferents coexisting with ART, 15 μL of 0.1 M EDTA, 15 μL of 0.1 mM a-ART, 15 μL of various concentrations of these ions and biomolecules and 95 μL of 1 mM VB1 were added sequentially into 860 μL Tris–HCl buffer solution. After incubation at 25 °C for 5 min, the fluorescence spectra were recorded.3. Results and discussion
3.1 UV-vis absorption and fluorescence response of ART to VB1
The UV-vis absorption spectra of ART, a-ART, VB1 and the VB1 reacted with a-ART were investigated. As shown in Fig. 1a, ART alone has no obvious absorption, while with the addition of 0.05 M NaOH to the ART solution, a new strong absorption peak located near 306 nm was found. This indicates that NaOH can catalyze the reaction of ART with H2O efficiently, forming a product with ultraviolet absorption, which is the basis of UV-vis spectrophotometry for detection of ART.31,32Fig. 1a shows that VB1 has two obvious UV absorption peaks at 225 and 265 nm, and upon addition of a-ART, the absorption band at 265 nm is markedly enhanced, revealing the reaction between VB1 and a-ART and the oxidized products formed. The absorption maximum for VB1–a-ART is slightly blue shifted from that of VB1 alone. This may be closed cycle of VB1 oxidized by a-ART.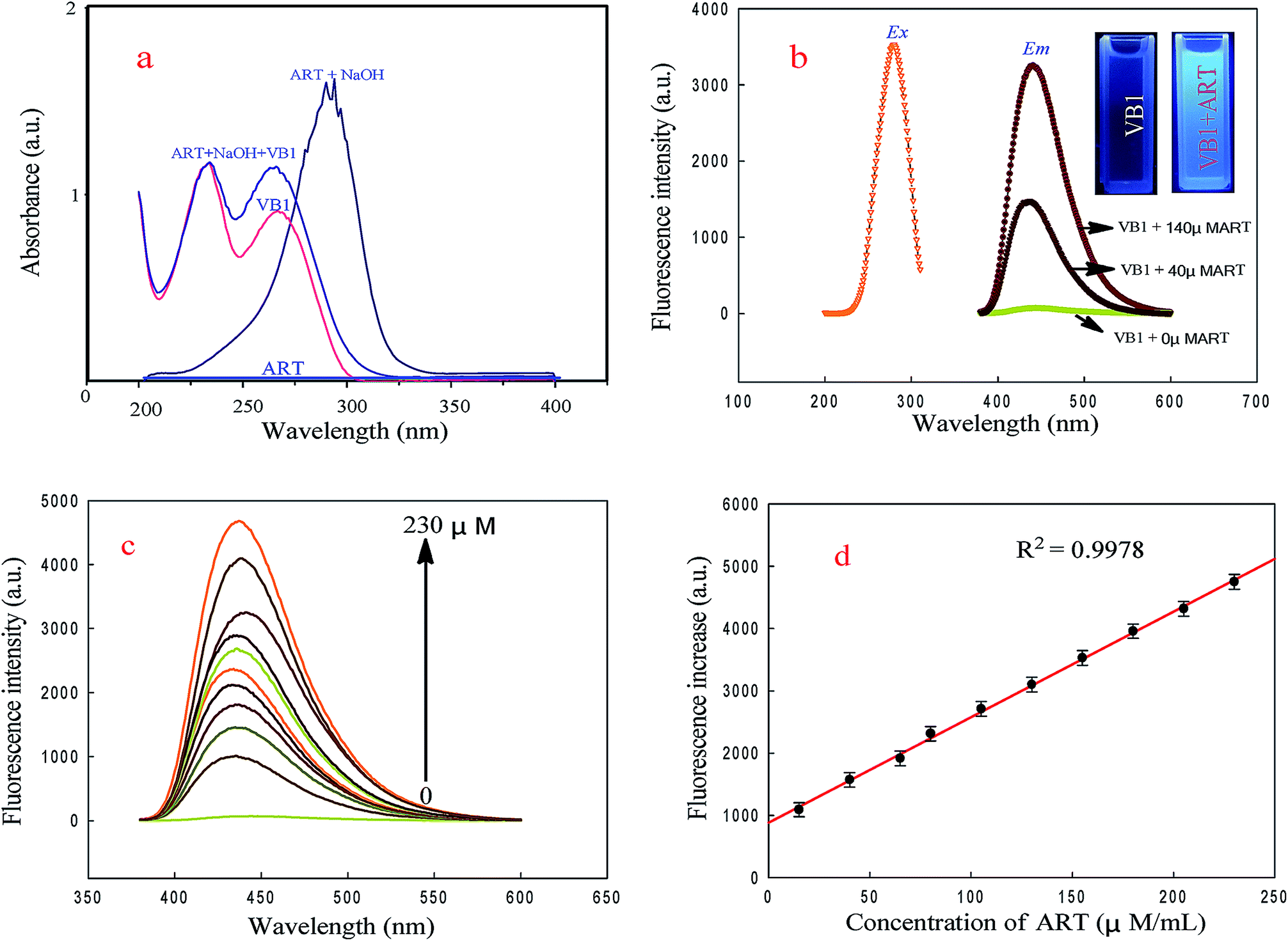 | ||
| Fig. 1 UV-vis spectra of ART, ART + NaOH, VB1 and VB1 in the presence of t a-ART (a); fluorescence excitation and emission spectra of VB1 solution and VB1 with the addition of 40 μM ART and 140 μM ART, respectively (b). Inset: photograph of VB1 solution and VB1 with the addition of a-ART under UV light of 254 nm; fluorescence intensity changes of the VB1 solutions in the presence of various concentrations of a-ART (c); the linear relationship of fluorescence increase of VB1versus the concentration of ART over the range from 1 to 230 μM.mL−1 (d). Conditions: VB1 concentration is 150 μM mL−1; Tris–HCl buffer (pH 7.5); excitation, 265 nm. | ||
Fluorescence studies (Fig. 1b) show that the maximum emission wavelength of VB1 appears at 442 nm under maximum excitation at 265 nm with the addition of a-ART; no fluorescence was found in the VB1 solution alone. Upon gradual addition of a-ART, the fluorescence intensity of VB1 solution at 442 nm was remarkably enhanced. The absolute quantum yield value of the product thiochromes was 0.453, which was measured according to the method mentioned in Section 1 in the ESI.† This reveals that the reaction of VB1 with a-ART happened, eliciting fluorescence turn-on (Fig. 1c). This result is consistent with UV-vis absorption spectra. Fig. 1d displays the relationship between the fluorescence intensity of VB1 and the concentration of a-ART, where the fluorescence intensity is increased with increasing concentration of a-ART.
3.2 Sensing mechanism for selective detection of ART using VB1
The principle of the a-ART–VB1 sensing system based on the oxidative cyclization of VB1 eliciting fluorescence turn-on is illustrated in Fig. 2. ART is a fat-soluble compound, while VB1 is water-soluble. It is difficult for them to react in the same solvent system. Additionally, there are no groups in ART for VB1 to bind to except for the endoperoxide group, which is a hindrance for the recognition of VB1. Fortunately, the lactone can be opened and release –OH and –COOH by hydrolysis. We have found that the hydrolysis of ART can not only solve the problem of solubility of ART in water, but also dramatically accelerates the conversion of VB1 into a fluorescent cyclic product. To confirm the presence of various functional groups in a-ART, a FT-IR experiment was carried out, and it suggested the characteristic absorption of –OH and –COOH stretching vibrations (Fig. 3a). Furthermore, the results of high performance liquid chromatography (Fig. 3b) validated the formation of hydrolyzates of ART.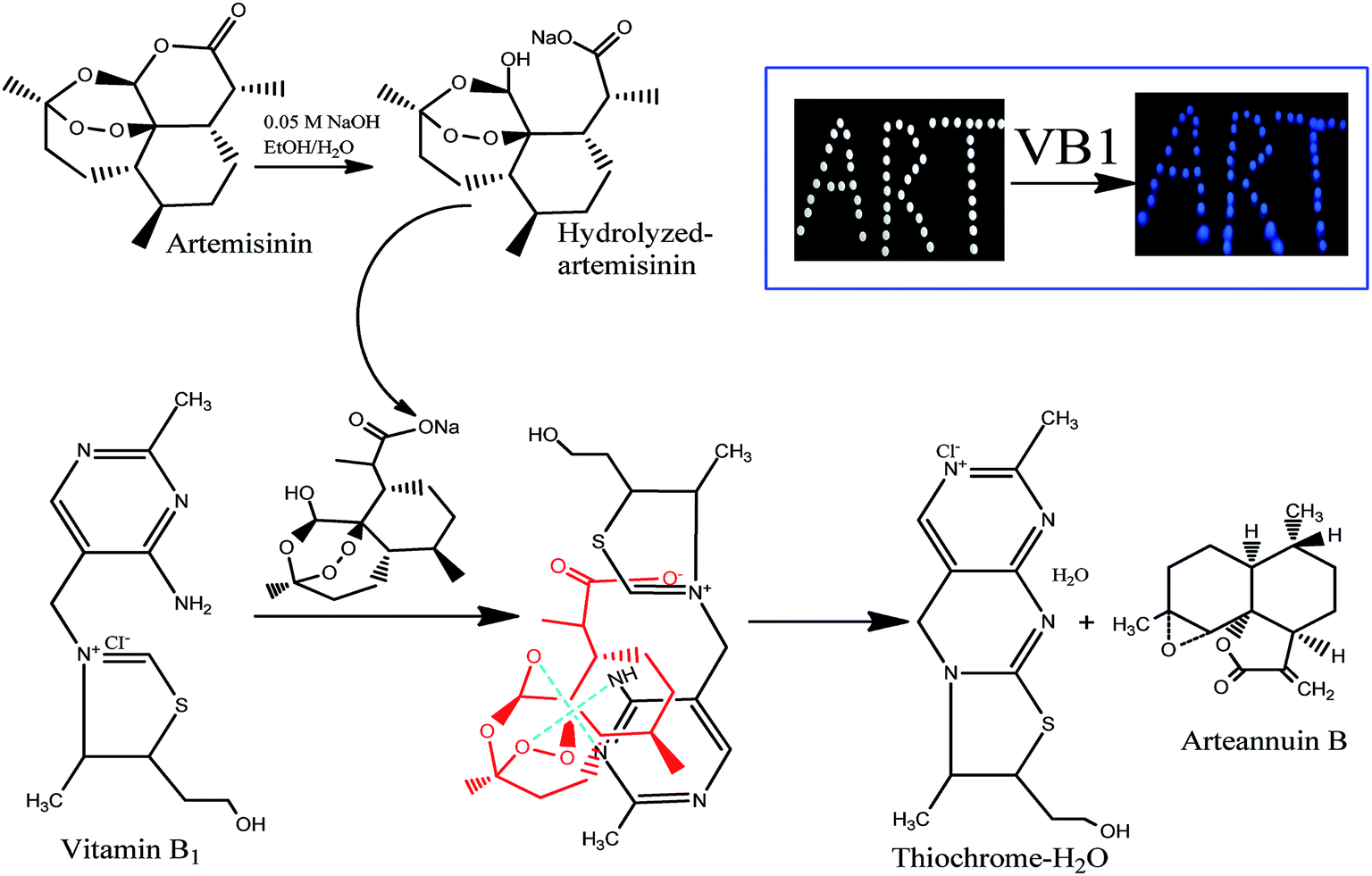 | ||
| Fig. 2 Schematic illustration for the hydrolysis of ART, reaction of the hydrolyzed ART with VB1 and selective detection of ART (inset: photograph of ART tablets pretreated with 0.05 M NaOH and dipped in a VB1 solution under UV light of 254 nm). | ||
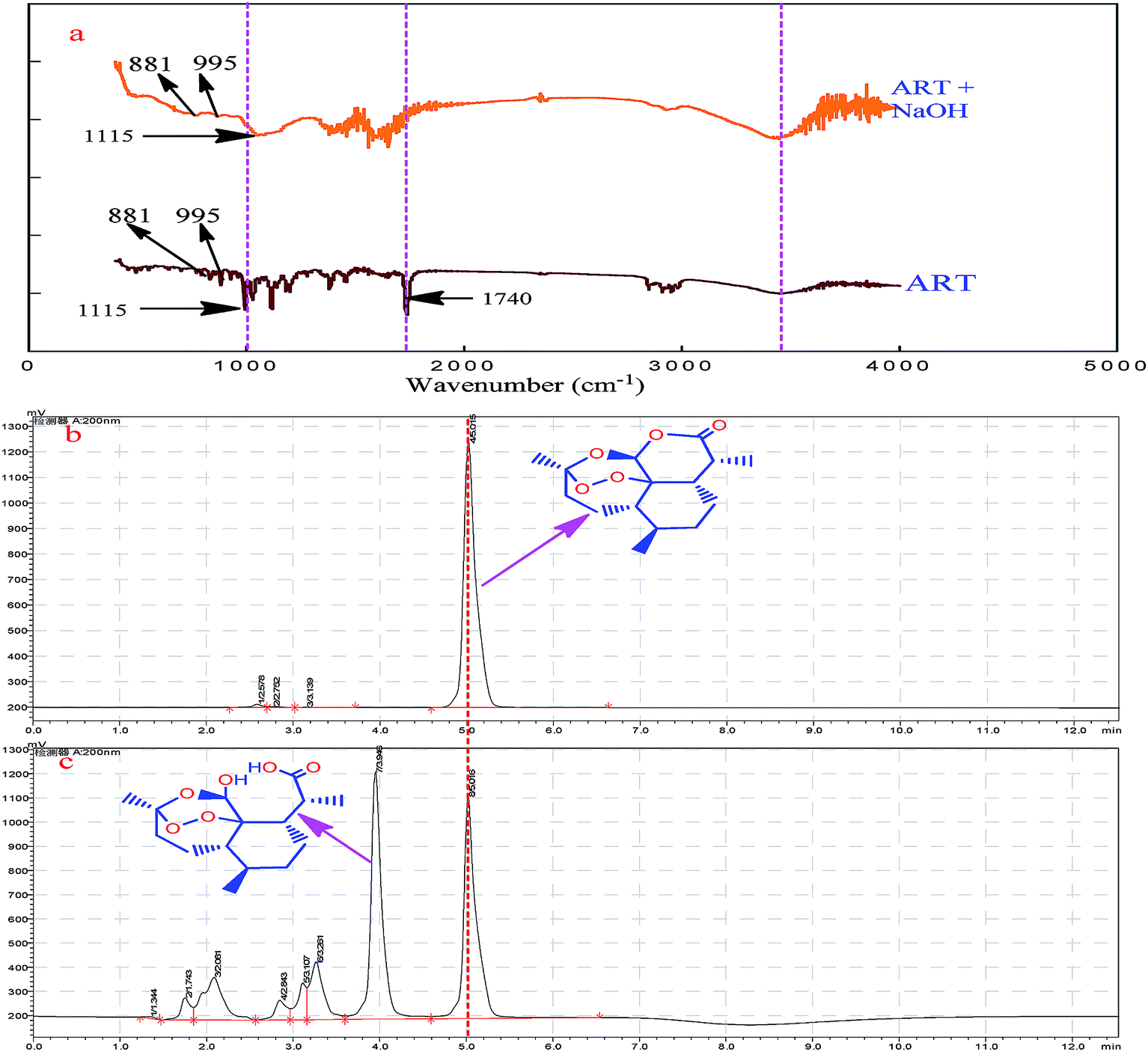 | ||
| Fig. 3 FTIR spectra of ART and the hydrolyzed ART (a); HPLC of ART (b) and the hydrolyzed ART (c), respectively. | ||
In this sensing system, a-ART binds to VB1via hydrogen bonds and forms an a-ART–VB1 supermolecule. As a result, the redox reaction of VB1 with a-ART becomes similar to the intramolecular reaction, enhancing the rate of reaction. Furthermore, VB1 exhibited good selectivity to a-ART over other substances, such as metal ions, amino acids and vitamin C. We speculate that this high selectivity of VB1 to a-ART may be related to the formation of a six membered-ring by the intramolecular hydrogen-bonding network and the electrostatic interaction between VB1 and a-ART. These results are in accordance with the UV absorption and fluorescence spectra, which clearly verified our proposed detection principle.
To confirm that the fluorescence sensing response of a-ART to VB1 is indeed due to the formation of thiochrome, mass spectrometry was used to investigate the reaction products of VB1 and a-ART. Into the a-ART solution (0.5 M L−1) in pH 7.5 Tris–HCl buffered water, VB1 solution (0.2 M L−1) in water was added under stirring; after a 5 min reaction the water was evaporated under vacuum, and the solid residue (reaction product) was then used for mass spectrometric measurement. The MS spectra for both ART and the reaction product are given in Fig. 4. It can be seen that the molecular weight (249) of the product of ART and the corresponding product equals the molecular weight of thiochrome (315) produced. This analysis strongly suggests that upon addition of VB1, the product has indeed resulted from the a-ART and VB1 and the fluorescence turn-on can be elicited.
 | ||
| Fig. 4 Mass spectra of ART (a) and the products in the reaction of a-ART with VB1 (b). | ||
3.3 Optimization of the VB1-based sensing method for ART detection
In order to obtain a better response of the fluorogenic probes, it is necessary to optimize the conditions that potentially affect the detection of ART, such as pH, concentration of VB1, reaction time and reaction temperature. As exhibited in Fig. S1a,† the emission intensity of the VB1–a-ART system showed only a slight increase as the pH value reached 7.5; the significant increase in fluorescence can be seen when the pH increases from 3.0 to 7.5. A possible reason is the pH effect on the reaction of VB1 with a-ART and the emission of thiochromes.33 In common plants, the microenvironmental pH value may vary from 4.0 to approximately 8.0. Therefore, we chose 7.5 as the pH value to evaluate the response of the sensor based on comprehensive consideration.For the sake of a good fluorescence response, an appropriate concentration of VB1 was needed. Taking into account the reaction of VB1 with ART and the characteristics of vulnerable oxidation of VB1 in air or light, the appropriate VB1 to ART mole ratio is 1.5:1.0 (Fig. S1b†). Response time is another key factor in evaluating the practical application of a probe. The response rate of the fluorescence signal of the VB1 to reaction time upon addition of a-ART was monitored subsequently. Fig. S1b† reveals that the fluorescence increase stops within 3 min and a further increase of time does not cause further enhancement of the fluorescence. This result demonstrates that the increase of VB1 in fluorescence intensity by a-ART is rapid and stable, implying a promising application for a fast and convenient sensing of a-ART without strict time control. Fig. S1c† shows that the optimum temperature for the reaction of a-ART with VB1 is 25 °C. The optimal experimental conditions were obtained when the pH of the sample solution was 7.5, reaction time was 5 min and concentration of VB1 was ∼460 μM at room temperature.
3.4 Selectivity and anti-interference performance of the sensing system
To assess the specificity of the VB1-based probe towards a-ART, the fluorescence response of the probe in the presence of interfering species including cysteine (Cys), lysine (Lys), Fe3+, Cu2+, Co2+, Zn2+, NO3−, PO43−, H2O2, ascorbic acid (VC), vitamin B6 (VB6), glucose and chlorophyll was recorded. The concentration of the amino acids Cys and Lys is 1.0 mM; for Zn2+, Co2+, Cu2+ and Fe3+, it is 0.1 mM; for glucose and chlorophyll, it is 10 μM; and for a-ART, it is 0.5 μM. And the comparison of the fluorescence intensities at 442 nm is given in Fig. 5a. It is obvious that only ART elicits significant fluorescence turn-on, while the addition of other compounds results in a negligible fluorescence change except in the case of chlorophyll, even though the concentration of these interferents is much higher than that of ART. These results demonstrate that the probe shows excellent selectivity toward ART over other competitive species. This is probably because a-ART has H-receptors and donors and can readily bind to VB1 by multiple hydrogen bonds.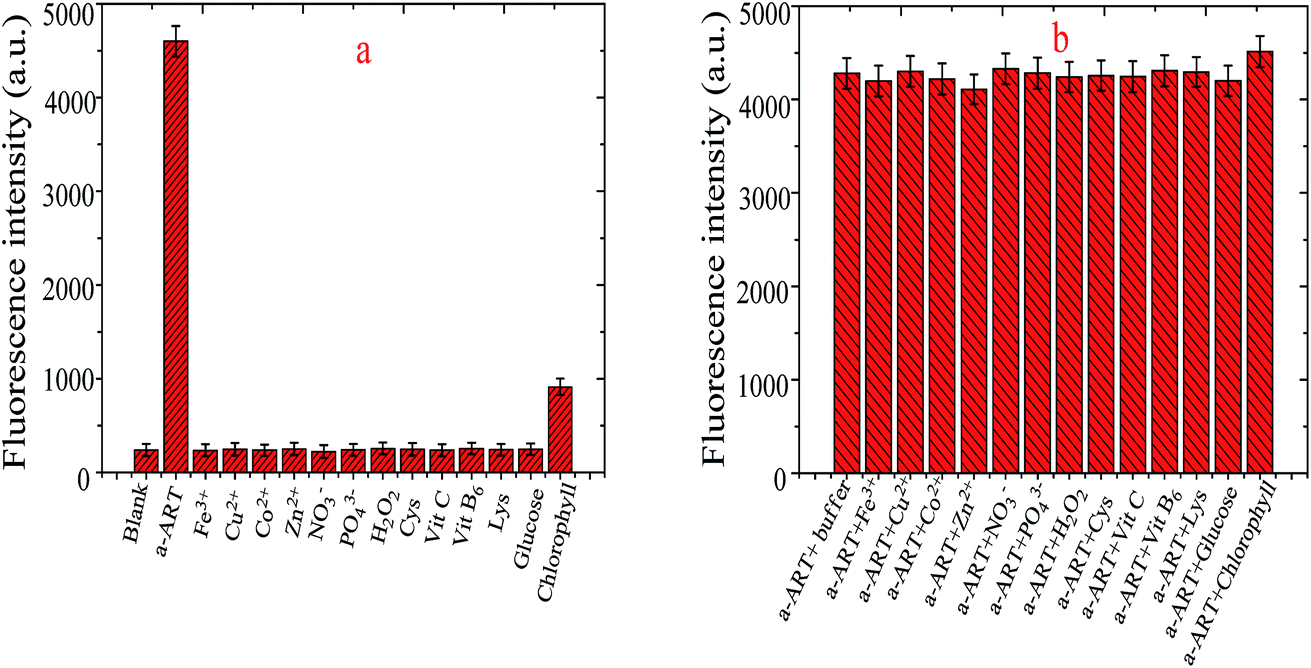 | ||
| Fig. 5 Fluorescence response of 10 mM VB1 after the addition of a-ART and other interfering substances separately. Blank represents the fluorescence intensity of VB1 in Tris–HCl buffer solution. The concentration of the interfering substances is 1 mM with the exception of chlorophyll, whose concentration is 10 μM. The concentration of a-ART is 5 μM (a). Fluorescence response of VB1 with the addition of a-ART and other interfering substances (b). The concentration of amino acids Cys and Lys is 1 mM; for Zn2+, Co2+, Cu2+ and Fe3+, it is 0.1 mM; for glucose and chlorophyll, it is 10 μM; and for a-ART, it is 0.5 μM. | ||
On the other hand, the interference of the above species was also investigated by studying their effects on the fluorescence increase of a-ART + VB1. Fig. 5b shows the fluorescence response of the probe towards a-ART in the presence of other competitive species. Moreover, the addition of the mixture incorporating metal ions and compounds results in a negligible change in the fluorescence intensity of VB1-based probes (data not shown). It is obvious that generally the co-existence of these species does not interfere either with the reaction of a-ART with the VB1-based probe or the subsequent fluorescence enhancement. These results suggest that the probe can function as a highly selective probe for ART.
3.5 Determination of ART using the VB1-based sensing method
As shown in Fig. 2d, we obtained a good linear relationship between concentration of ART and fluorescence intensity. The linear range is from 1.0 μM to 230 μM with a correlation coefficient of 0.9978 (n = 10). The calibration equation is shown in eqn (1):| ΔF = 6.38 × 10−5 + 3.72 × 10−6 C | (1) |
ART is naturally present in the leaves of A. annua L. but the content of ART varies with different varieties, harvesting methods and production areas. For A. annua L. utilized in the industry, there are different plucking times, different places of origin and different parts of the plant that are used, and the content of ART might vary significantly in different places of origin and different parts of the plant which may affect the therapeutic effects and safety. To evaluate the applicability and reliability of the VB1-based probe for ART determination, we applied the developed method for the analysis of ART in mature stem and leaf of A. annua L. The proposed VB1-based fluorogenic probes were applied for quantitative determination of ART in various dried leaf samples of A. annua L. from Yongzhou (China). The content (mean ± SD) of ART in dried leaf samples is shown in Table 1. The triplicate determination results of each compound in each sample all showed good consistency with outstanding RSD values (all less than 2.8%).
| Samplesa | Calculated concentration (ng g−1) | RSDb (%) |
|---|---|---|
| a The samples were provided by Yongzhou Xincheng Biotechnolgy Co. Ltd. The test solutions were obtained from the stock solution prepared as described in the Experimental section. b Mean ± RSD of six measurements. | ||
| S01 | 23.1 ± 0.173 | 2.38 |
| S02 | 18.7 ± 0.196 | 2.74 |
| S03 | 21.3 ± 0.207 | 2.12 |
| S04 | 17.6 ± 0.182 | 2.80 |
| S05 | 18.8 ± 0.201 | 1.97 |
| S06 | 19.8 ± 0.189 | 2.04 |
| S07 | 18.4 ± 0.178 | 2.66 |
| S08 | 15.4 ± 0.193 | 2.51 |
| S09 | 17.7 ± 0.158 | 2.36 |
| S10 | 18.2 ± 0.184 | 1.97 |
| S11 | 19.5 ± 0.193 | 1.84 |
| S12 | 22.1 ± 0.196 | 2.79 |
Recovery experiments were also conducted in naphthoquine phosphate tablet samples to estimate the feasibility of the VB1-based fluorogenic probe through the standard addition method. The ART content of naphthoquine phosphate tablet samples was found to be 9.6 mM. The determined ART concentrations were in good agreement with the corresponding actual ART content (10 mM). Four known amounts of standard ART were added into the real samples and the recoveries were examined. As shown in Table 2, recoveries of 104.0%, 101.7%, 99.1% and 99.8% for naphthoquine phosphate tablets were obtained when 30, 60, 120, and 240 mM ART were added. RSDs are less than 3.5%. These results indicate the practicability of the proposed VB1-based sensing platform and the proposed method is promising for accurate measurement of ART in real samples.
3.6 Comparison of the VB1-based sensing system with the reported methods
The potentiality of our proposed fluorogenic probes is compared with the reported methods for ART determination. The linearity range and LOD for ART determination using VB1-based fluorogenic probes are compared with those using HPLC, UV-vis spectrophotometry, cyclic voltammetry, and nanomaterial-based electrochemical methods, presented in Table 3. It shows that a wide linear range and better LOD were obtained with our proposed method compared to the reported methods. These gratifying results can be attributed to the inherent characteristics of VB1-based fluorogenic probes. However, coupling of chemiluminescence (CL) and the molecular imprinting method with a graphene modified electrode showed a little more sensitivity compared to the present method. Thus, the VB1-based method is found to be simple, rapid and low cost compared to chromatographic, voltammetric, CL, nanomaterial-based electrochemical and conventional UV-vis methods.| Analytical methods | ART | Samples | References | |
|---|---|---|---|---|
| Linearity range | LOD | |||
| UV-vis absorption | 2.0–10.0 μg mL−1 | 0.2451 μg mL−1 | Tablets | 34 |
| Indirect UV-vis absorption | 2.0–80 μg mL−1 | 0.30 μg mL−1 | Tablets | 35 |
| Chemiluminescence | 0.01–0.068 μM mL−1 | 3.2 nM mL−1 | Human plasma | 36 |
| Indirect voltammetry | 2.0–200 μM mL−1 | 0.8 μM mL−1 | Tablets | 37 |
| LC/MS | 10–140 μg mL−1 | 50 ng mL−1 | Extract | 38 |
| HPLC-DAD | 177.0–3541.9 μM mL−1 | 1.8 μM mL−1 | Fresh leaves and human serum | 39 |
| Nanosensor (CV detection) | 0.01–0.08 μg mL−1 | 0.0035 μg mL−1 | Extract | 23 |
| Imprinted sensor (CV detection) | 0.01–0.4 μM mL−1 | 2.0 nM mL−1 | Tablets | 18 |
| Present method | 1–230 μM mL−1 | 11.5 nM mL−1 | — | |
4. Conclusions
A simple and effective strategy for detecting ART based on the fluorescence determination of thiochromes generated from the selective oxidation of VB1 by ART has been established. This VB1-based sensing methodology avoids tedious synthesis for the preparation of specific ART probes and makes the fluorescence determination of ART easy to realize, and is more simple and sensitive than the reported electrochemical methods. Furthermore, superior selectivity of VB1 to ART and good water-solubility of ART can be achieved by hydrolyzing ART. The results of the recovery test showed that the proposed method can detect ART in tablets with high recoveries. The methods developed for detecting ART are facile, economic, and environmentally friendly. Moreover, this method may be potentially extended to track other endoperoxides and ART derivatives. The finding that ART can react with thiamine has the potential to be exploited for developing other sensors, such as electrochemical sensors.Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
This research was supported by the National Natural Science Foundation of China (No. 31527803 and 21545010) and the Natural Science Foundation of State Key Laboratory of Life Organic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences (No. SKLBNPC17344). The authors also express their gratitude for the financial support from the project of Hunan Natural Science Research Fund (No. 2018JJ2422) and National Natural Science Foundation of China (No. 11405013).References
- Y. Dieye, B. Mbengue, S. Dagamajalu, M. M. Fall, M. F. Loke, C. M. Nguer, A. Thiam, J. Vadivelu and A. Dieye, PeerJ, 2016, 4, e1965 CrossRef PubMed.
- R. E. Desjardins, C. J. Canfield, J. D. Haynes and J. D. Chulay, Antimicrob. Agents Chemother., 1979, 16, 710–718 CrossRef CAS.
- P. Phukon, K. Radhapyari, B. K. Konwar and R. Khan, Trans. R. Soc. Trop. Med. Hyg., 2004, 98, 318–321 CrossRef.
- G. O. Gbotosho, T. M. Okuboyejo, C. T. Happi and A. Sowunmi, Asian Pac. J. Trop. Dis., 2011, 1, 195–202 CrossRef.
- E. Leonardi, G. Gilvary, N. J. White and F. Nosten, Trans. R. Soc. Trop. Med. Hyg., 2001, 95, 182–183 CrossRef CAS.
- W. R. J. Taylor and N. J. White, Antimalarial drug toxicity, Drug Saf., 2004, 27, 25–61 CrossRef CAS.
- L. Li, D. Pabbisetty, P. Carvalho, M. A. Avery, J. S. Williamson and B. A. Avery, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2008, 867, 131–137 CrossRef CAS.
- N. Lindegardh, J. Tarning, P. V. Toi, T. T. Hien, J. Farrar, P. Singhasivanon, N. J. White, M. Ashton and N. P. Day, J. Pharm. Biomed. Anal., 2009, 49, 768–773 CrossRef CAS.
- T. Gordi, E. Nielsen, Z. Yu, D. Westerlund and M. Ashton, J. Chromatogr. B: Biomed. Sci. Appl., 2000, 742, 155–162 CrossRef CAS.
- H. Liu, Q. Li, S. Li, Y. Zou and A. Gu, J. Chromatogr. Sci., 2008, 46, 122–126 CAS.
- A. A. Lapkin, A. Walker, N. Sullivan, B. Khambay, B. Mlambo and S. Chemat, J. Pharm. Biomed. Anal., 2009, 49, 908–915 CrossRef CAS PubMed.
- C. A. Peng, J. F. Ferreira and A. J. Wood, J. Chromatogr. A, 2006, 1133, 254–258 CrossRef CAS.
- A. Amponsaa-Karikari, N. Kishikawa, Y. Ohba, K. Nakashima and N. Kuroda, Biomed. Chromatogr., 2006, 20, 1157–1162 CrossRef CAS PubMed.
- J. Suberu, L. Song, S. Slade, N. Sullivan, G. Barker and A. A. Lapkin, J. Pharm. Biomed. Anal., 2013, 84, 269–277 CrossRef CAS.
- A. Carra, R. Bagnati, R. Fanelli and M. Bonati, Food Chem., 2014, 142, 114–120 CrossRef CAS.
- X. Wang, X. E. Zhao, B. Yang, H. Dong, D. Liu and L. Huang, Phytochem. Anal., 2011, 22, 280–284 CrossRef CAS.
- X. Yang, T. Gan, X. Zheng, D. Zhu and K. Wu, Bull. Korean Chem. Soc., 2008, 29, 1386–1390 CrossRef CAS.
- J. Xie, C. Cai, H. Yang and X. Chen, Anal. Lett., 2013, 46, 107–119 CrossRef.
- X. Dong, N. Wang, S. Wang, X. Zhang and Z. Fan, J. Chromatogr. A, 2004, 1057, 13–19 CrossRef CAS.
- I. S. Lee and C. D. Hufford, Pharm. Res., 1990, 48, 345–355 CAS.
- F. C. Gong, Z. D. Xiao, Z. Cao and D. X. Wu, Talanta, 2007, 72, 1453–1457 CrossRef CAS.
- P. Phukon, K. Radhapyari, B. K. Konwar and R. Khan, Mater. Sci. Eng., C, 2014, 37, 314–320 CrossRef CAS.
- N. Adányi, K. Majer-Baranyi, M. Berki, B. Darvas, B. Wang, I. Szendrő and A. Székács, Sens. Actuators, B, 2017, 239, 413–420 CrossRef.
- X. F. Yang, T. Gan, X. J. Zheng, D. Z. Zhu and K. B. Wu, Bull. Korean Chem. Soc., 2008, 29, 1386–1390 CrossRef CAS.
- D. N. Shetty, B. Narayana and S. Samshuddin, J. Chem. Pharm. Res., 2012, 4, 1647–1653 CAS.
- Y. Hu, S. Chen, Y. Han, H. Chen, Q. Wang, Z. Nie, Y. Huang and S. Yao, Chem. Commun., 2015, 51, 17611–17614 RSC.
- G. Strack, S. Chinnapareddy, D. Volkov, J. Halámek, M. Pita, I. Sokolov and E. Katz, J. Phys. Chem. B, 2009, 113, 12154–12159 CrossRef CAS.
- X. Xi, J. Li, H. Wang, Q. Zhao and H. Li, Microchim. Acta, 2015, 182, 1273–1279 CrossRef CAS.
- M. M. Shahid, P. Rameshkumar and N. M. Huang, Microchim. Acta, 2016, 183, 911–916 CrossRef.
- R. P. Brinas, M. H. Hu, L. P. Qian, E. S. Lymar and J. F. Hainfeld, J. Am. Chem. Soc., 2008, 130, 975–982 CrossRef CAS.
- H. N. Eisohly, E. M. Croom and M. A. Elsohly, Pharm. Res., 1987, 4, 258–260 CrossRef.
- J. Hong, J. Maguhn, D. Freitag and A. Kettrup, J. Anal. Chem., 1998, 361, 124–128 CAS.
- M. Wada, K. Inoue, A. Ihara, N. Kishikawa, K. Nakashima and N. Kuroda, J. Chromatogr. A, 2003, 987, 189–195 CrossRef CAS.
- E. Emmanuel, U. Aniefiok, J. Ekarika, U. Anwanabasi and E. Etienne, J. Pharm. Innov., 2016, 5, 04–07 Search PubMed.
- E. E. Attih, C. O. Usifoh and H. O. Olandimeji, Bull. Environ., Pharmacol. Life Sci., 2015, 4, 90–99 Search PubMed.
- L. Zehnacker, M. C. Nevers, V. Sinou, D. Parzy, C. Creminon, D. Parzy and S. Azoulay, Anal. Bioanal. Chem., 2015, 407, 7823–7830 CrossRef CAS PubMed.
- C. Wang, Y. T. Zholudov, A. Nsabimana, G. Xu and J. Li, Anal. Chim. Acta, 2016, 937, 39–42 CrossRef CAS.
- J. Jastrebova, L. Nyholm, K. E. Markides and Y. Bergqvist, Analyst, 1998, 123, 313–317 RSC.
- H. Liu, O. Li, S. Li, Y. Zou and A. Gu, J. Chromatogr. Sci., 2008, 46, 22–126 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ay02312c |
| This journal is © The Royal Society of Chemistry 2019 |