
Amino-functionalized mesoporous silica nanospheres (MSN-NH2) as sorbent for extraction and concentration of synthetic dyes from foodstuffs prior to HPLC analysis†
Peige
Qin
,
Yixin
Yang
,
Wenqi
Li
,
Jing
Zhang
,
Qian
Zhou
and
Minghua
Lu
 *
*
Institute of Environmental and Analysis Science, School of Chemistry and Chemical Engineering, Henan University, Kaifeng 475004, Henan, China. E-mail: mhlu@henu.edu.cn
First published on 26th November 2018
Abstract
In this work, amino-functionalized mesoporous silica nanospheres (MSN-NH2) were synthesized and demonstrated as useful sorbents of dispersive solid-phase extraction (dSPE) for extraction and preconcentration of synthetic dyes (tartrazine, amaranth, sunset yellow, allura red, and orange II) from foodstuffs prior to high-performance liquid chromatography (HPLC) analysis. The MSN-NH2 was characterized by transmission electron microscopy, Fourier-transform infrared spectroscopy, nitrogen adsorption and desorption isotherms, and elemental analysis. To achieve optimized extraction efficiency, several critical factors including amount of MSN-NH2, sample pH, adsorption time, adsorption temperature, desorption time, and desorption solvents, were systematically investigated. Under the optimized conditions, the enrichment factors were higher than 120. The limits of detection were found to be 0.10–0.30 ng mL−1. The method showed excellent linearity in the range of 0.45–1000.0 ng mL−1 with correlation coefficients (R2) ≥ 0.9951. The intra-day and inter-day precision (n = 5) were 1.72–3.64% and 1.45–4.52%. The recoveries at three concentration levels ranged from 82.7 to 116.3%. The proposed dSPE by using MSN-NH2 as sorbent coupled with HPLC was successfully applied to the analysis of synthetic dyes in real samples.
1. Introduction
Compared with natural dyes that are usually associated with instability and easy degradation during the manufacturing process, synthetic dyes are more widely used in the food industry because of their low price, high effectivity, and good stability.1 However, some synthetic dyes, especially azo-dyes, could be converted into harmful derivatives in the human body.2,3 More and more studies have revealed that many of these synthetic dyes can increase human health risks including allergic and asthmatic reactions, DNA damage, hyperactivity, and carcinogenesis.4–8 Thus, the use of synthetic dyes must be strictly monitored to assure food safety and consumer health. Therefore, it is very important to establish an efficient and convenient method for the determination of synthetic dyes in foodstuffs.Nowadays, many of the techniques and methods including electrochemical sensors,9–11 spectrometric methods,2,12 and chromatographic methods13 were developed for the analysis of synthetic dyes in various foodstuffs. Due to simple, economical and reliable properties, high-performance liquid chromatography (HPLC) was most commonly preferred among these methods.14–17 HPLC with an ultraviolet (UV) or diode array detector (DAD) usually suffers from low detection sensitivity. To overcome this drawback, HPLC coupled with a highly sensitive detector (e.g. mass spectrometry, MS) has been typically adopted in the determination of synthetic dyes.18–23 Although LC-MS methods possess high sensitivity and selectivity for the determination of synthetic dyes in food samples, they are often not utilized in small institutes and factories since they not only require a high cost for the instrument and its maintenance, but also need professional technicians. So, a highly efficient sample preparation technique coupled with HPLC may have more practical application in routine analysis.
In recent years, various sample preparation methods have been developed for the extraction and preconcentration of synthetic dyes in different food matrices. Sha24et al. reported an aqueous two-phase approach based on ionic liquid liquid–liquid microextraction (LLME) of five synthetic food colorants. Bazregar25et al. developed in-tube electro-membrane extraction that involved sub-microliter organic solvent consumption for synthetic food dyes. Qi26et al. proposed a solid-phase extraction (SPE) procedure by using a nanofiber mat as sorbent after solvent extraction and freeze-salting out for the purification of anionic and cationic dyes. Among them, SPE has become a widely used technique due to the simplicity and flexibility of the approach, and variety of available analytes.27 The choice of effective sorbent materials is the most important factor for obtaining higher enrichment efficiency in SPE procedures. Recently, dispersive solid-phase extraction (dSPE) has earned more attention because it combines the advantages of liquid–liquid extraction and SPE, such as sufficient contact time and more choice of sorbents. Thus far, many kinds of materials are potential candidates for dSPE to extract and preconcentrate target analytes, such as graphitized multi-walled carbon nanotubes,28 mesoporous carbon,29 CdFe2O4 microspheres,30 and crown ether coated multi-walled carbon nanotubes (MWCNTs).26 Among these materials, it is important to find a highly selective and effective dSPE sorbent for the analysis of synthetic dyes.
Due to high specific surface area, large pore volume, as well as controllable and uniform particle size, mesoporous silica nanospheres (MSN) have attracted considerable attention since their development by Kresge in 1900s.31 Moreover, MSN are easily grafted with suitable organic groups because of the large amount of hydroxyl groups on the surface. MSN have gained a wide range of promising applications, particularly in drug,32 gene,33 and protein34 deliveries because of their good biocompatibility. In recent years, MSN or functionalized MSN have been used as an ideal material to remove heavy metals (e.g., Fe2+, Cu2+ and U(VI))35,36 and microcystins37 from water resources.
In this study, the investigation of amino-functionalized mesoporous silica nanospheres (MSN-NH2) for efficient dSPE, and high-performance liquid chromatography (HPLC) analysis of synthetic dyes in different foodstuffs is reported for the first time. Five compounds including tartrazine, amaranth, sunset yellow, allura red, and orange II containing sulfonic groups (see Fig. S1 in ESI†) were selected as target analytes to evaluate the extraction performance of this method. Factors related to the sample preparation performance were studied and the validation of the method was in terms of linearity, limits of detection and quantification, precision, reproducibility, and recovery. Finally, the proposed method was successfully used for extraction and preconcentration of synthetic dyes in real samples.
2. Experimental
2.1 Reagents and standards
Cetyltrimethylammonium bromide (CTAB, 99%), (3-aminopropyl) trimethoxysilane (APTMS), tetraethyl orthosilicate (TEOS, 98%), succinic anhydride (SA), N,N-dimethylformamide (DMF), and ammonium acetate (CH3COONH4) were purchased from Aladdin Industrial Corporation (Shanghai, China). HPLC grade methanol and acetone were supplied by Merck (Darmstadt, Germany). Ammonium nitrate (NH4NO3) and ethanol were obtained from Tianjin Kermel Chemical Reagent Co., Ltd. (Tianjin, China). Ultrapure water (18.2 MΩ) used throughout the study was produced by a Millipore Milli-Q ultrapure water system (Millipore, Bedford, MA, USA). All the chemicals were used without further purification. Analytical standards including tartrazine, amaranth, sunset yellow, allura red and orange II were supplied by Aladdin Industrial Corporation (Shanghai, China). The stock standard solution containing each analyte with a concentration of 5.0 μg mL−1 was prepared in ultrapure water. The working standard solutions (0.0001–1 μg mL−1) were prepared by serial dilutions of the stock standard solution with ultrapure water. The stock standard solution and working standard solution were stored in amber glass volumetric flasks in the dark at 4 °C. All solvents and solutions for HPLC analysis were filtered through a Millipore filter (pore size 0.45 μm).2.2 Instrumentation
The morphological analysis of the synthesized functionalized MSN was performed using a JEM 2100 transmission electron microscope (TEM, Tokyo, Japan). Infrared (IR) spectra of MSN and MSN-NH2 were recorded on a VERTEX 70 Fourier transform infrared (FT-IR) spectrometer (Bruker, Karlsruhe, Germany) in the wavelength range of 4000–500 cm−1. The nitrogen adsorption and desorption isotherms of functionalized MSN were measured using an ASAP 2020 (Micromeritics, USA). Elemental analysis was performed on a Vario EL elemental analyzer with CHNS/O mode (Elementar Co., Germany).2.3 Synthesis of functionalized MSN
2.4 dSPE procedures and HPLC analysis
The proposed dSPE methodology was performed as follows: initially, 12.0 mg of MSN-NH2 sorbent was directly added into a 50 mL centrifuge tube that contained 10 mL of mixed standard working solution with a concentration of 500 ng mL−1 for each analyte. The mixture was sonicated for 12 min and then centrifuged for 5 min at 8000 rpm. After that, the supernatant was discarded and the residual solution with MSN-NH2 was transferred to a 5.0 mL centrifuge tube. Similarly, the supernatant was discarded, and the sorbents were collected. The target analytes were eluted from the MSN-NH2 by dispersion of sorbents into 1 mL of acetone solution containing 20% pH = 11 B–R buffer solution with sonication for 20 min. The eluate was collected and concentrated to dryness under a mild nitrogen stream and re-dissolved in 50.0 μL water. Finally, after filtration through 0.45 μm membrane, 5 μL of the solution was injected into the HPLC system for analysis.The synthetic dyes were separated and determined using an Agilent 1260-HPLC system equipped with an Eclipse Plus C18 column (4.6 × 100 mm, 3.5 μm, Agilent Technologies, Calif., USA). Methanol (B) and 0.02 M aqueous ammonium acetate solution (A) were used as the mobile phase with the gradient elution programs as follows: 0–6.5 min, 15–32% B; 6.5–10 min, 32% B; 10–14 min, 32–15% B. The flow rate was 0.5 mL min−1. The column temperature was maintained at 25 °C, and the injection volume was 5 μL. Two different wavelengths were selected for detection and quantification of the synthetic dyes (254 nm for tartrazine, amaranth, sunset yellow, and allura red; 485 nm for orange II).
2.5 Sample preparation
The carbonated beverage (orange flavor), candy (corn flavor), ice cream and canned yellow peaches were purchased from a local supermarket and stored in a refrigerator. The carbonated beverage was diluted by ultrapure water with the volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, and then mixed for 10 min with a sonication bath at room temperature to remove the CO2 gas. Then the pH was adjusted to 4.0 with 0.1 mol L−1 NaOH or HCl. Afterwards, the beverage solution was centrifuged for 5 min at 8000 rpm and the supernatant was collected by filtering through a 0.45 μm filter. The solid samples (candy, ice cream and canned yellow peach) were homogenized before weighing. Afterwards, exactly 1.0 g of each sample was weighed out and dissolved in 20 mL of ultrapure water at pH 4.0, heated to 60 °C for 5 min with stirring, and cooled; then the solution was centrifuged (10 min, 6000 rpm) to remove the insoluble particles. The filtrate was collected for further enrichment process.
:1, and then mixed for 10 min with a sonication bath at room temperature to remove the CO2 gas. Then the pH was adjusted to 4.0 with 0.1 mol L−1 NaOH or HCl. Afterwards, the beverage solution was centrifuged for 5 min at 8000 rpm and the supernatant was collected by filtering through a 0.45 μm filter. The solid samples (candy, ice cream and canned yellow peach) were homogenized before weighing. Afterwards, exactly 1.0 g of each sample was weighed out and dissolved in 20 mL of ultrapure water at pH 4.0, heated to 60 °C for 5 min with stirring, and cooled; then the solution was centrifuged (10 min, 6000 rpm) to remove the insoluble particles. The filtrate was collected for further enrichment process.
2.6 The buffer of desorption solvent
The Britton–Robinson Buffer solution (B–R buffer) of various pH was prepared by adding different volumes of NaOH (0.2 mol L−1) to 0.04 mol L−1 of (H3PO4–HAc–H3BO3) mixed solution.3. Results and discussion
3.1 Characterization of synthetic materials
The morphological features of the synthesized MSN, MSN-COOH, MSN-NH2 are shown in Fig. 1. It can be seen from Fig. 1A–C that the MSN, MSN-COOH and MSN-NH2 nanoparticles were found to possess spherical particles with an average particle size of about 30 nm. Meanwhile, the TEM images also confirmed the existence of highly ordered pores on the synthetic materials surface.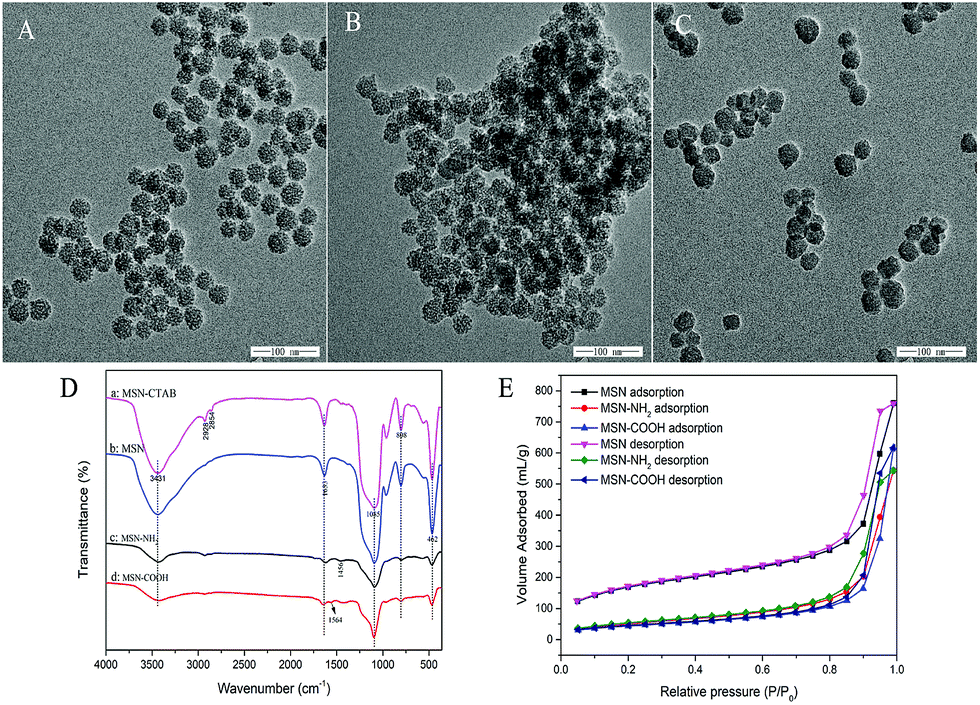 | ||
| Fig. 1 TEM image of MSN (A), MSN-COOH (B) and MSN-NH2 (C); FT-IR images of MSN-CTAB, MSN, MSN-NH2 and MSN-COOH nanospheres (D); N2 adsorption–desorption isotherms of MSNs and functionalized MSN nanospheres (E). | ||
The FT-IR spectra of MSN, MSN-NH2 and MSN-COOH are presented in Fig. 1D. The FT-IR spectra of pristine MSNs and all functionalized MSNs showed absorption peaks of silanol groups at around 1085 cm−1, 808 cm−1, and 462 cm−1 which are attributed to Si–O bonds. In the spectrum of MSN-CTAB, the absorption peak at 2928 cm−1 and 2854 cm−1 are related to C–H stretching vibrations, which disappeared in the MSN spectrum after the template was removed. MSN was reported to exhibit strong absorption bands at about 3431 cm−1 and 1633 cm−1 (MSN spectrum), which are associated with the deformation vibration of interlayer water and –OH stretching vibrations of silanol groups, respectively. From Fig. 1D, the much wider and broader OH peak of MSN-NH2 may be attributed to the effect of amino groups. Additionally, the absorption bands at 2928 cm−1 and 1456 cm−1 of MSN-NH2 correspond to the C–H bond and the N–H bond, respectively. The bands appearing close to 1564 cm−1 are assigned to the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bands. These results indicate that functionalized MSN were successfully prepared.
O bands. These results indicate that functionalized MSN were successfully prepared.
Further evidence can also be found with the elemental analysis of MSN and MSN-NH2, as shown in Table S1 (ESI†). In comparison with MSN, the percentage of N present in MSN-NH2 increases from 0 to 2.82%. Meanwhile, the percentages of C and H elements incorporated in the amino-functionalized MSN (MSN-NH2) are 8.54% and 1.174%, respectively. The elemental analysis further confirmed the successful synthesis of MSN-NH2.
The adsorption property of functionalized MSN was characterized via nitrogen gas porosity measurements. Fig. 1E shows the N2 adsorption–desorption isotherms. MSNs and functionalized MSNs all exhibited typical type IV curves with a sharp uptake at a high relative pressure (P/P0 > 0.8), which demonstrated that all the samples are mesoporous materials. The corresponding data obtained from the experimental isotherms are exhibited in Table S2 (ESI†). After modification of MSNs with functionalized groups, the amount of adsorbed nitrogen and mean diameter decrease, indicating that the pores were plugged after the grafting procedure. In addition, the calculated results in Table S2† show the same decrease in surface area, pore volume and pore size after the grafting process. This was most probably because the functionalized groups can easily fill the uniform micropores and thus restrict the access of nitrogen into the pores.
3.2 Comparison of adsorption performance of MSN and functionalized MSNs
The adsorption performance of MSNs, MSN-COOH, and MSN-NH2 nanospheres were investigated and the results are presented in Fig. 2. It can be seen that the MSN-NH2 exhibited the best adsorption performance compared with those of MSN and MSN-COOH. The probable reason can be attributed to the surface of MSN-NH2 containing a high concentration of amino groups, which can form strong interactions with hydroxyl groups and/or sulfonic acid groups of synthetic dyes.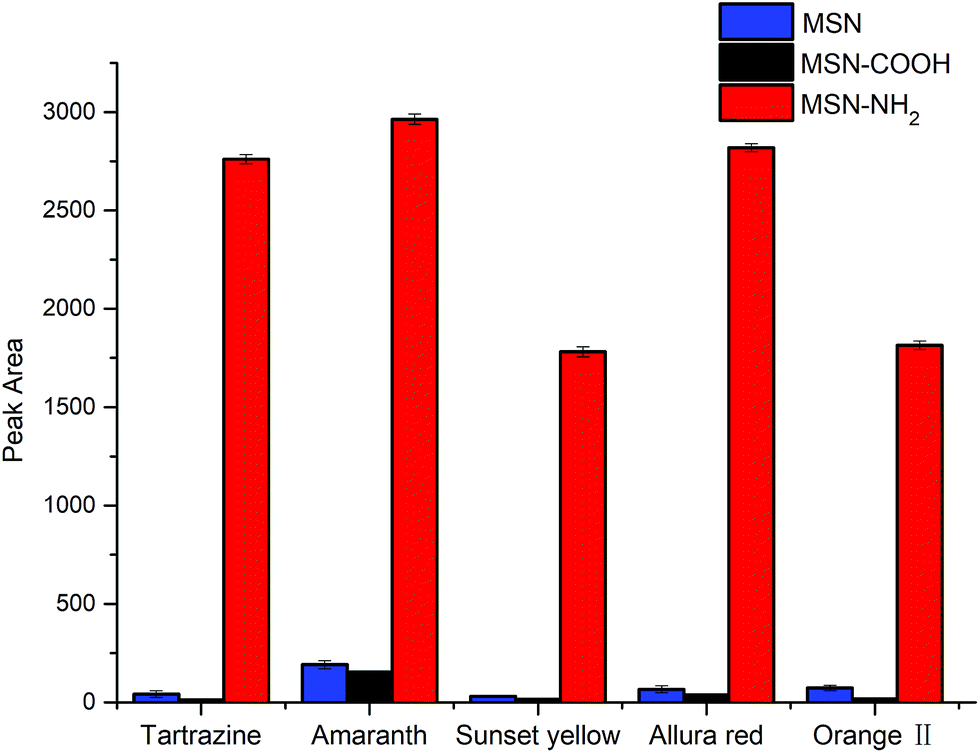 | ||
| Fig. 2 Comparison of adsorption performance of MSN and functionalized MSN for target analytes. Conditions: amount of sorbent, 15 mg; adsorption temperature, room temperature; extraction time, 20 min; desorption time, 25 min; desorption solvent, acetone: pH = 9 B–R buffer (v/v = 8:2); analyte concentration, each at 500 ng mL−1. | ||
3.3 Optimization of extraction
10 mL sample solution with a concentration of 500 ng mL−1 for each synthetic dye was selected to evaluate the performance of MSN-NH2 as the sorbent of dSPE. In order to obtain the optimum extraction efficiency, several parameters including amount of sorbent, adsorption temperature, adsorption time, and sample pH were investigated.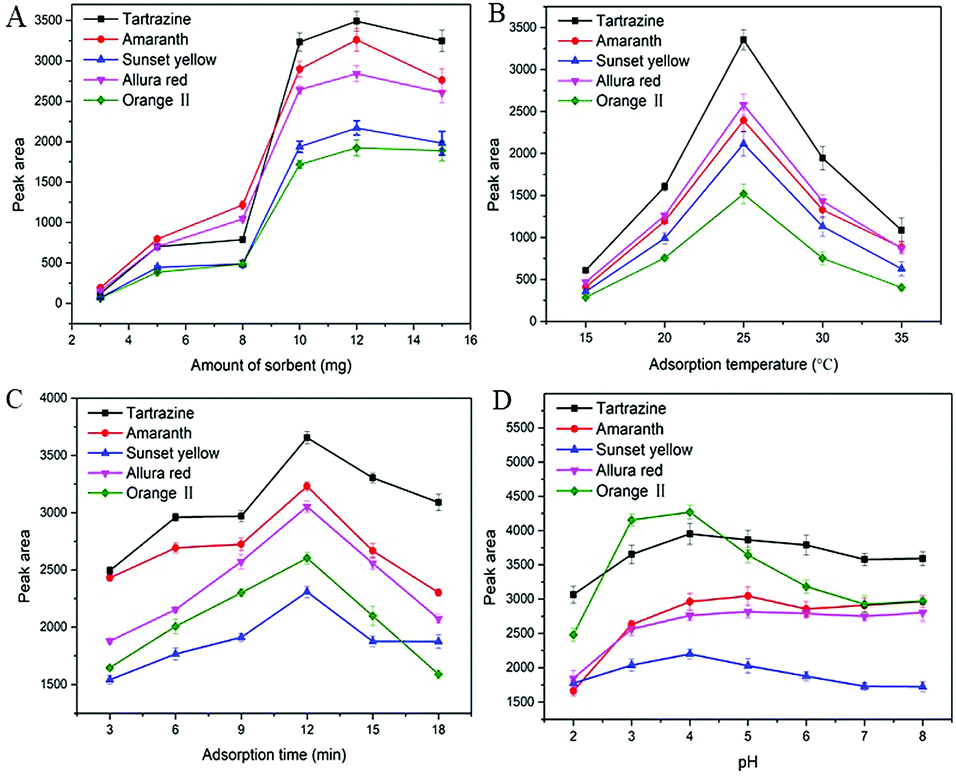 | ||
| Fig. 3 Effects of amount of sorbent (A), adsorption temperature (B), adsorption time (C) and pH (D) on the adsorption efficiency. | ||
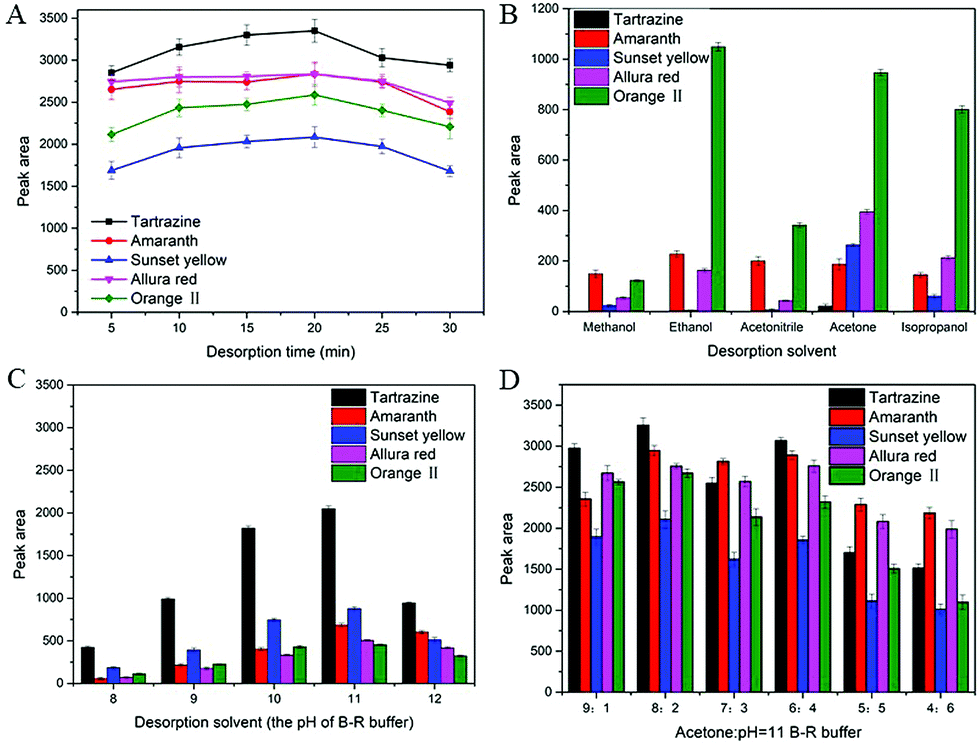 | ||
| Fig. 4 Effect of desorption time (A), desorption solvent (organic solvent) (B), desorption solvent (the pH of B–R buffer) (C) and desorption solvent (acetone: pH = 11 B–R buffer) (D) on the desorption efficiency. | ||
3.4 Optimization of desorption
To further improve the desorption efficiency, different proportions of acetone and B–R buffer (pH = 11) solution mixture as the eluting solution was evaluated. As shown in Fig. 4D, the acetone/B–R buffer (pH = 11) solution (8:2, v/v) provided the highest peak area for all synthetic dyes than that of other solvents. Thus, acetone/B–R buffer (pH = 11) solution (8:2, v/v) was selected as the optimized desorption solvent.
3.5 Validation of the HPLC-DAD method
Some quantitative parameters of the proposed dSPE-HPLC method were established including linearity, limits of detection (LODs, S/N = 3), limits of quantification (LOQs, S/N = 10), enrichment factors (EFs), precision (relative standard deviations, RSDs), and reproducibility under the optimized conditions. The results are summarized in Table 1. The calibration curves were plotted by the peak area versus the target analyte concentration. A good linearity was obtained for all target analytes, with correlation coefficients (R2) higher than 0.9951. The LODs for analytes were calculated to be from 0.10 to 0.30 ng mL−1, which were much lower than those obtained without enrichment by the MSN-NH2 nanoparticles. The LOQs obtained from the lowest concentration of linearity, ranged from 0.45 to 2.0 ng mL−1. The enrichment factor (EF) was calculated based on the following equation:| EF = Smethod/Sdirect |
| Analytes | Linear range (ng mL−1) | R 2 | LODs (ng mL−1) | LOQs (ng mL−1) | EFs | Repeatability, RSD (%) | |||
|---|---|---|---|---|---|---|---|---|---|
| Before | After | Intra-day (n = 5) | Inter-day (n = 5) | Batch to batch (n = 3) | |||||
| Tartrazine | 0.60–1000 | 0.9973 | 25 | 0.15 | 0.60 | 157 | 2.32% | 1.73% | 2.71% |
| Amaranth | 0.45–1000 | 0.9958 | 20 | 0.10 | 0.45 | 143 | 2.87% | 4.52% | 1.63% |
| Sunset yellow | 0.80–1000 | 0.9989 | 40 | 0.25 | 0.80 | 151 | 3.46% | 1.45% | 3.81% |
| Allura red | 0.75–1000 | 0.9976 | 30 | 0.25 | 0.75 | 146 | 1.72% | 1.55% | 4.39% |
| Orange II | 2.00–1000 | 0.9951 | 50 | 0.30 | 2.00 | 120 | 3.64% | 2.11% | 4.77% |
| Samples | Analytes | Found | Recovery mean (%) ± SD | ||
|---|---|---|---|---|---|
| Level 1 | Level 2 | Level 3 | |||
| a ND: not detected. The samples were spiked at 20 ng mL−1 (level 1), 50 ng mL−1 (level 2), and 200 ng mL−1 (level 3) with solution containing tartrazine, amaranth, sunset yellow, allura red, and orange II. | |||||
| Corn flavored candy | Tartrazine | 1.26 ± 0.02 μg g−1 | 96.7 ± 6.7 | 110.9 ± 4.3 | 82.6 ± 3.5 |
| Amaranth | ND | 108.5 ± 2.6 | 80.0 ± 4.8 | 81.7 ± 4.2 | |
| Sunset yellow | ND | 114.7 ± 4.8 | 95.0 ± 3.8 | 87.0 ± 3.7 | |
| Allura red | ND | 89.6 ± 5.2 | 84.3 ± 3.5 | 106.7 ± 4.1 | |
| Orange II | ND | 116.3 ± 6.1 | 85.6 ± 5.2 | 87.8 ± 3.6 | |
| Ice cream | Tartrazine | 1.52 ± 0.05 μg g−1 | 110.9 ± 3.9 | 109.5 ± 3.5 | 104.6 ± 2.4 |
| Amaranth | 1.66 ± 0.12 μg g−1 | 106.6 ± 2.5 | 110.0 ± 3.9 | 115.7 ± 3.4 | |
| Sunset yellow | 1.08 ± 0.17 μg g−1 | 93.4 ± 4.1 | 80.6 ± 2.8 | 94.5 ± 2.7 | |
| Allura red | ND | 82.7 ± 3.8 | 108.6 ± 3.7 | 85.7 ± 3.2 | |
| Orange II | ND | 110.7 ± 3.5 | 82.3 ± 4.3 | 90.5 ± 4.2 | |
| Carbonated beverage (orange) | Tartrazine | 3.00 ± 0.01 μg mL−1 | 116.3 ± 3.8 | 85.4 ± 2.7 | 107.7 ± 3.2 |
| Amaranth | ND | 92.5 ± 4.2 | 84.5 ± 3.6 | 112.7 ± 2.6 | |
| Sunset yellow | 3.90 ± 0.02 μg mL−1 | 89.1 ± 4.7 | 106.0 ± 4.5 | 83.3 ± 3.9 | |
| Allura red | ND | 108.6 ± 2.5 | 83.1 ± 2.3 | 86.0 ± 3.2 | |
| Orange II | ND | 83.5 ± 3.6 | 91.2 ± 3.9 | 90.0 ± 4.1 | |
| Canned yellow peaches | Tartrazine | ND | 93.4 ± 4.3 | 98.1 ± 2.4 | 90.4 ± 2.6 |
| Amaranth | ND | 88.3 ± 2.9 | 82.3 ± 4.7 | 83.3 ± 3.8 | |
| Sunset yellow | ND | 116.8 ± 3.2 | 94.1 ± 2.7 | 93.0 ± 2.3 | |
| Allura red | ND | 110.6 ± 2.4 | 98.2 ± 3.3 | 89.7 ± 3.2 | |
| Orange II | ND | 89.2 ± 3.2 | 89.3 ± 4.2 | 94.5 ± 4.3 | |
3.6 Real samples analysis
To further evaluate the performance of the developed method for analysis of synthetic dyes, four different foodstuffs including an orange flavored beverage, corn flavored candy, ice cream, and canned yellow peaches were selected. The real samples were prepared as described in Section 2.5. The chromatograms for the analysis of corn flavored candy are shown in Fig. 5A. It can be seen from Fig. 5A that the interference peaks were eliminated, and the target peak was most enhanced with MSN-NH2 as clean and preconcentration materials. The target peak (tartrazine) was further confirmed by the analysis of a spiked and standard sample. The content of tartrazine was calculated as 1.26 μg g−1 in corn flavored candy. Fig. 5B shows the chromatograms for the analysis of ice cream with the developed method. Three target analytes including tartrazine, amaranth, and sunset yellow were detected with corresponding concentrations of 1.516, 1.661, and 1.083 μg g−1, respectively. The method was also applied to the analysis of orange flavored beverage and canned yellow peaches (chromatograms are not presented here). The content of all detected target analytes are listed in Table 2.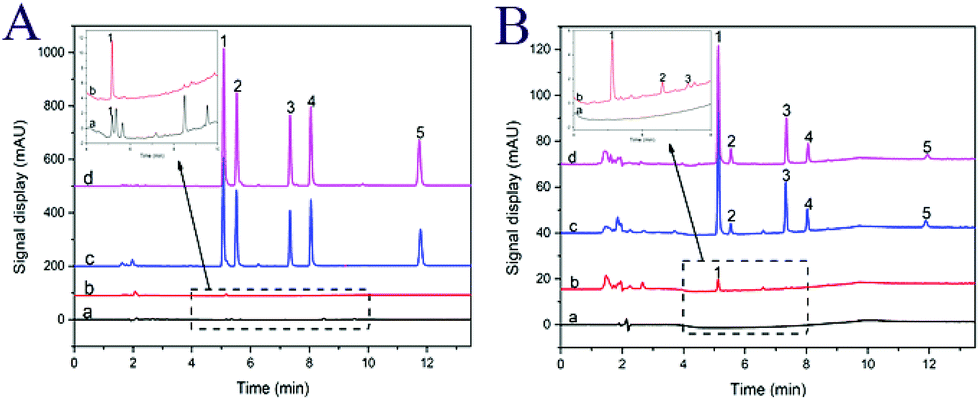 | ||
| Fig. 5 HPLC-DAD chromatograms of target analytes in real samples. (A) Corn flavored candy sample before (a) and after (b) enrichment with MSN-NH2; spiked sample (c) and standard solution (d) with a concentration of 200 ng mL−1 by MSN-NH2 enrichment. (B) Ice cream sample before (a) and after (b) enrichment with MSN-NH2; spiked sample (c) and standard solution (d) with a concentration of 20 ng mL−1 by MSN-NH2 enrichment. Peaks: (1) tartrazine, (2) amaranth, (3) sunset yellow, (4) allura red, (5) orange II. | ||
To further evaluate method accuracy, the recoveries for the analysis of synthetic dyes were investigated by spiking standard solution into the food samples at three different concentrations (20, 50, and 200 ng mL−1). For each concentration level, three replicate experiments were performed. As shown in Table 2, the determined recoveries of the synthetic dyes were in the range of 82.3–116.8% with RSDs between 2.3 and 5.2%, respectively.
3.7 Reusability of the sorbents
The reusability and stability of MSN-NH2 nanospheres as the sorbent of dSPE were investigated by six adsorption/desorption (regeneration) cycles using the same batch of MSN-NH2 particles. Before each cycle, the sorbent was rinsed three times with desorption solvent to remove the residual eluent. The results (Fig. S2 in ESI†) indicated that the extraction efficiency of sorbent was almost maintained in the first four cycles. After the sixth recycle, the extraction efficiency was still higher than 71% for all target analytes. These data demonstrated that the MSN-NH2 particles had good reusability and great potential for recycling use.3.8 Comparison with other methods
The performance of the proposed method was evaluated by comparing with other reported methods for extraction of synthetic dyes. As can be seen from Table 3, this method exhibited relatively lower LODs and wider linear range compared with other reported methods.3,40–43 These data indicated that the MSN-NH2 have good absorption ability for synthetic dyes, and the developed method can be applied to the determination of analytes in food samples.| Methoda | Sorbentb | LOD (ng mL−1) | Linear range (ng mL−1) | Recovery (%) | Ref. |
|---|---|---|---|---|---|
| a PMME, polymer monolith microextraction; MSPE, magnetic solid-phase dispersion extraction; MSPD, matrix solid phase dispersion; d-μ-SPE, dispersive solid-phase microextraction; CE, capillary electrophoresis. b dASNPs, diamino moiety functionalized silica nanoparticles; NH2-LDC-MP, amino-functionalized low degrees of cross-linking magnetic polymer. | |||||
| PMME-HPLC-DAD | Al2O3 | 9.5–9.9 | 50–5000 | 90.4–109.2 | 42 |
| MSPE-HPLC | Fe3O4@rGO | 10.02–15.91 | 50–10000 |
88.95–95.89 | 43 |
| MSPD coupled to UPLC-MS/MS | — | 7.01–200 | 5–500 | 81.0–122.0 | 44 |
| d-μ-SPE-CE | dASNPs | 30–72 | 160–5000 | 82.7–114.6 | 45 |
| MSPE-LC-MS | NH2-LDC-MP | 0.45–1.25 | 2.5–1000 | 84.0–116.2 | 3 |
| dSPE-HPLC-DAD | MSN-NH2 | 0.15–0.30 | 0.45–1000 | 80.6–116.3 | This work |
4. Conclusions
In this study, amino-functionalized mesoporous silica nanospheres (MSN-NH2) were prepared and used as the sorbent of dSPE for extraction and preconcentration of synthetic dyes. The as-prepared MSN-NH2 exhibited large specific surface area, high water-stability, as well as good biocompatibility and chemical stability. The materials demonstrated excellent adsorption efficiency toward synthetic dyes since the –NH2 group on the surface of MSN-NH2 particles could interact with hydroxyl group and/or sulfonic acid group of synthetic dyes. By using MSN-NH2 as a highly efficient sorbent of dSPE, a low-cost and sensitive HPLC method for the determination of synthetic dyes was developed. Since low values of LODs and LOQs could be obtained via HPLC-DAD, this relatively inexpensive technique could be applied in screening synthetic dyes in foodstuffs to ensure food safety.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This study was supported by the National Nature Science Foundation of China (21477033); the Program for Science & Technology Innovation Talents in Universities of Henan Province (17HASTIT003); the Program for Development in Science and Technology of Henan Province (172102310608) are gratefully acknowledged.References
- C. C. Qin, W. L. Guo, Y. Liu, Z. C. Liu, J. Qiu and J. B. Peng, Food Anal. Methods, 2017, 10, 2293–2301 CrossRef.
- T. I. Tikhomirova, G. R. Ramazanova and V. V. Apyari, Food Chem., 2017, 221, 351–355 CrossRef CAS PubMed.
- X.-H. Chen, Y.-G. Zhao, H.-Y. Shen, L.-X. Zhou, S.-D. Pan and M.-C. Jin, J. Chromatogr. A, 2014, 1346, 123–128 CrossRef CAS PubMed.
- P. Amchova, H. Kotolova and J. Ruda-Kucerova, Regul. Toxicol. Pharmacol., 2015, 73, 914–922 CrossRef CAS PubMed.
- P. Mpountoukas, A. Pantazaki, E. Kostareli, P. Christodoulou, D. Kareli, S. Poliliou, C. Mourelatos, V. Lambropoulou and T. Lialiaris, Food Chem. Toxicol., 2010, 48, 2934–2944 CrossRef CAS PubMed.
- Y. F. Sasaki, S. Kawaguchi, A. Kamaya, M. Ohshita, K. Kabasawa, K. Iwama, K. Taniguchi and S. Tsuda, Mutat. Res., Genet. Toxicol. Environ. Mutagen., 2002, 519, 103–119 CrossRef CAS.
- A. Benvidi, S. Abbasi, S. Gharaghani, M. D. Tezerjani and S. Masoum, Food Chem., 2017, 220, 377–384 CrossRef CAS PubMed.
- J. Y. Shin and M. Y. Jung, J. Agric. Food Chem., 2017, 65, 9917–9923 Search PubMed.
- S. Chandran, L. A. Lonappan, D. Thomas, T. Jos and K. G. Kumar, Food Anal. Methods, 2014, 7, 741–746 CrossRef.
- P. Sierra-Rosales, C. Toledo-Neira and J. A. Squella, Sens. Actuators, B, 2017, 240, 1257–1264 CrossRef CAS.
- Q. Cheng, S. Xia, J. Tong and K. Wu, Anal. Chim. Acta, 2015, 887, 75–81 CrossRef CAS PubMed.
- E. Heidarizadi and R. Tabaraki, Talanta, 2016, 148, 237–246 CrossRef CAS PubMed.
- M. Kucharska and J. Grabka, Talanta, 2010, 80, 1045–1051 CrossRef CAS PubMed.
- J. H. Chen, G. M. Zhou, H. Y. Qin, Y. Gao and G. L. Peng, Anal. Methods, 2015, 7, 6537–6544 RSC.
- T. Rejczak and T. Tuzimski, Food Anal. Methods, 2017, 10, 3572–3588 CrossRef.
- J. Zhang, J. B. Shao, P. Guo and Y. M. Huang, Anal. Methods, 2013, 5, 2503–2510 RSC.
- W. d. A. Siqueira Bento, B. P. Lima and A. P. S. Palm, Food Chem., 2015, 183, 154–160 CrossRef PubMed.
- F. Martin, J.-M. Oberson, M. Meschiari and C. Munari, Food Chem., 2016, 197, 1249–1255 CrossRef CAS PubMed.
- C.-F. Tsai, C.-H. Kuo and D. Y.-C. Shih, J. Food Drug Anal., 2015, 23, 453–462 CrossRef CAS PubMed.
- P. Qi, Z.-h. Lin, G.-y. Chen, J. Xiao, Z.-a. Liang, L.-n. Luo, J. Zhou and X.-w. Zhang, Food Chem., 2015, 181, 101–110 CrossRef CAS PubMed.
- X. Q. Li, Q. H. Zhang, K. Ma, H. M. Li and Z. Guo, Food Chem., 2015, 182, 316–326 CrossRef CAS PubMed.
- J. Li, X.-M. Ding, D.-D. Liu, F. Guo, Y. Chen, Y.-B. Zhang and H.-M. Liu, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2013, 942, 46–52 CrossRef PubMed.
- H. G. Gao, W. J. Gong and Y. G. Zhao, Anal. Sci., 2015, 31, 205–210 CrossRef CAS PubMed.
- O. Sha, X. Zhu, Y. Feng and W. Ma, Food Chem., 2015, 174, 380–386 CrossRef CAS PubMed.
- M. Bazregar, M. Rajabi, Y. Yamini, A. Asghari and Y. A. Asl, J. Chromatogr. A, 2015, 1410, 35–43 CrossRef CAS PubMed.
- F. Qi, N. Jian, L. Qian, W. Cao, Q. Xu and J. Li, Anal. Bioanal. Chem., 2017, 409, 5697–5709 CrossRef CAS PubMed.
- L.-J. Du, Y.-H. Hu, Q.-Y. Wang, Q.-D. Zhang, Y.-B. Chen, L.-Q. Peng, S.-L. Pan, Q. Li and J. Cao, Food Chem., 2018, 262, 118–128 CrossRef CAS PubMed.
- S.-L. Wang, X.-Q. Pang, J. Cao, W. Cao, J.-J. Xu, Q.-Y. Zhu, Q.-Y. Zhang and L.-Q. Peng, J. Chromatogr. A, 2015, 1418, 12–20 CrossRef CAS.
- N. Yahaya, M. M. Sanagi, T. Mitome, N. Nishiyama, W. A. W. Ibrahim and H. Nur, Food Anal. Methods, 2015, 8, 1079–1087 CrossRef.
- D. Zhang, L. Zhang and T. Liu, Microchim. Acta, 2018, 185, 248–253 CrossRef.
- C. Kresge, M. Leonowicz, W. Roth, J. Vartuli and J. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- F. Liu, J. Wang, Q. Cao, H. Deng, G. Shao, D. Y. B. Deng and W. Zhou, Chem. Commun., 2015, 51, 2357–2360 RSC.
- D. Niu, Z. Liu, Y. Li, X. Luo, J. Zhang, J. Gong and J. Shi, Adv. Mater., 2014, 26, 4947–4953 CrossRef CAS PubMed.
- A. K. Meka, P. L. Abbaraju, H. Song, C. Xu, J. Zhang, H. Zhang, M. Yu and C. Yu, Small, 2016, 12, 5169–5177 CrossRef CAS PubMed.
- W. Naowanon, R. Chueachot, S. Klinsrisuk and S. Amnuaypanich, Powder Technol., 2018, 323, 548–557 CrossRef CAS.
- B. Wang, Y. Zhou, L. Li and Y. Wang, Sci. Total Environ., 2018, 626, 219–227 CrossRef CAS PubMed.
- W. Teng, Z. Wu, D. Feng, J. Fan, J. Wang, H. Wei, M. Song and D. Zhao, Environ. Sci. Technol., 2013, 47, 8633–8641 CrossRef CAS.
- P. G. Qin, Y. X. Yang, X. T. Zhang, J. H. Niu, H. Yang, S. F. Tian, J. H. Zhu and M. H. Lu, Nanomaterials, 2018, 8, 14, DOI:10.3390/nano8010004.
- M. P. Daryasari, M. R. Akhgar, F. Mamashli, B. Bigdeli and M. Khoobi, RSC Adv., 2016, 6, 105578–105588 RSC.
- Y. Fan, M. Zhang and Y. Q. Feng, J. Chromatogr. A, 2005, 1099, 84–91 CrossRef CAS PubMed.
- J. A. Barreto, R. Dos Santos de Assis, R. J. Cassella and V. A. Lemos, Talanta, 2019, 193, 23–28 CrossRef CAS PubMed.
- W. J. Li, X. Zhou, S. S. Tong and Q. Jia, Talanta, 2013, 105, 386–392 CrossRef CAS.
- X. Wang, N. Chen, Q. Han, Z. Yang, J. Wu, C. Xue, J. Hong, X. Zhou and H. Jiang, J. Sep. Sci., 2015, 38, 2167–2173 CrossRef CAS.
- H. Li, N. Sun, J. Zhang, S. Liang and H. Sun, Anal. Methods, 2014, 6, 537–547 RSC.
- F. J. Liu, C. T. Liu, W. Li and A. N. Tang, Talanta, 2015, 132, 366–372 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ay02215a |
| This journal is © The Royal Society of Chemistry 2019 |