
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Addressing the practicalities of anodic stripping voltammetry for heavy metal detection: a tutorial review
Alexandra J.
Borrill†
 ab,
Nicole E.
Reily†
ac and
Julie V.
Macpherson
*a
ab,
Nicole E.
Reily†
ac and
Julie V.
Macpherson
*a
aDepartment of Chemistry, University of Warwick, Coventry CV4 7AL, UK. E-mail: j.macpherson@warwick.ac.uk
bDiamond Science and Technology Centre for Doctoral Training, UK
cNatural Environment Research Council, Central England NERC Training Alliance Doctoral Training Program, UK
First published on 15th October 2019
Abstract
Anodic Stripping Voltammetry (ASV) has the capability to detect heavy metals at sub ppb-level with portable and cheap instrumentation making it ideal for in the field (at the source) analysis, however, commercial activity is surprisingly limited. The more commonly used liquid mercury electrodes are now obsolete due to toxicity concerns, and replacements are all based around solid electrodes, which come with their own challenges. This tutorial review aims to discuss the experimental practicalities of ASV, providing a clear overview of the issues for consideration, which can serve as a guide for anyone wanting to undertake analytical ASV. Choice of electrode material (with or without subsequent modification) and solution composition (pH, electrolyte, buffer) are important parameters, as well as an understanding of pH dependent metal speciation and possible intermetallic effects. Measurements made on model solutions often differ from those made on environmental samples with the latter containing organic matter, biological and inorganic species, which themselves can adsorb metal ions. Consideration should also be given to the method of solution collection and the sample container utilised. ASV can be a powerful tool to an analytical chemist, however optimisation for the application of interest is essential, which this review aims to help guide.
1. Introduction
Heavy metals are defined as naturally occurring elements with a high atomic weight and density, at least five times greater than water.1 A number of heavy metals are found on the Environmental Quality Standards Directive List including As, Cd, Cu, Cr, Fe, Pb, Hg, Ni and Zn, which are highlighted as priority substances for assessment of water quality.2 Whilst some heavy metals are essential to life in trace quantities, they can be toxic at higher concentrations.3,4 Others in comparison are toxic even in small quantities.5–7 Identification and detection is thus essential, down to very low concentrations, sub-ppb. Although heavy metals occur naturally within the environment anthropogenic activities have led to an increase in their abundance within the natural system,1 with water bodies and sediments often acting as sinks for pollutants. Some heavy metals bioaccumulate up the food chain so analysis of primary sources is necessary for entire ecosystem health.8 Quantitative heavy metal analysis is not only important in environmental systems but in pharmaceuticals,9–11 food stuffs,12,13 and biological samples such as blood, urine and human hair.12,14–17Importantly, the speciation of a metal controls its bioavailability and therefore toxicity. Compounds only pose a risk if they are able to enter cells, which is the case for free (hydrated) metal ions and lipid soluble complexes.18–21 Metals that are strongly bound to ligands or inorganic particles (and are not lipid soluble) are often considered non-toxic or inert. Labile metal complexes have weakly coordinating ligands; the more labile the metal the higher the rate of ligand exchange.
Inductively Coupled Plasma-Mass Spectrometry (ICP-MS) and ICP-Optical Emission Spectrometry (OES) are the main techniques used for heavy metal detection. Whilst both can detect ppb concentrations, ICP-MS instruments are capable of ppt detection limits.22–24 For analysis, these techniques require samples to be processed and analysed in a laboratory. The instrumentation is large, expensive and requires a trained operative. Such methods are not easily adaptable to “at the source” measurements. Prior to analysis, the solution is typically strongly acidified (<pH 2), forcing all metal ions, whether strongly bound or not, into the free state. Solid samples are acid digested to form acidic solutions prior to analysis.
In contrast Anodic Stripping Voltammetry (ASV) offers less complex, lower cost and smaller footprint instrumentation with similar sub-ppb limits of detection (LOD).25,26 It has thus long been proposed as a viable technique for at the source or on-line heavy metal detection.21,27–29 ASV has been shown to be capable of detecting up to thirty different elements,30,31 although some e.g. As, require more involved detection protocols than others.32 Whilst the bulk of analysable species for ASV are metals, cathodic stripping voltammetry and adsorptive stripping voltammetry (not discussed herein) can be used to determine concentrations of organic and inorganic compounds such as sulfides, thiols, halides,30,33 pesticides and pharmaceuticals.34,35 In contrast to ICP techniques, unless the solution is deliberately acidified, ASV provides information on the concentration of free/labile metal at the measurement pH of the solution. Interestingly, despite substantial literature on the subject little commercial activity using ASV has emerged, suggesting the methodology is challenging to implement. This tutorial review aims to address why this is by developing an understanding of the factors involved and the challenges associated with ASV.
1.1. History
Polarography, the precursor technique to modern voltammetry, was initially developed in 1922 by Heyrovský, who was awarded the Nobel Prize in Chemistry in 1959 for his work.36 Polarography uses a dropping mercury electrode (DME) to monitor the change in current upon application of a voltage. Using polarography, metal ion detection down to 10−7–10−8 M was possible through measurement of the cathodic currents associated with metal ion reduction.37,38 This led to early commercial Hg-based instrumentation.39 In 1931, Zbinden reported the use of “stripping analysis” for metal detection with proof-of-concept studies focused on Cu.40,41 Historically, Hg was the working electrode of choice for ASV in a DME or thin film format.31,42 However, as Hg now features in the top ten chemicals of major public health concern43 it is no longer commercially viable as an electrode material and all existing Hg-based products are being phased out in accordance with the 2013 Minamata Convention.44 Thus there was a need to find alternative electrode materials for ASV with many papers dedicated to the testing of new electrodes in an attempt to find materials that rival liquid Hg (section 3.2).33,362. Electrochemical fundamentals
ASV involves a two-step process. First cathodic reduction of a labile or free metal ion to its zero-valence metallic state on the electrode surface, a process known as electrodeposition, eqn (1), Fig. 1. Deposition is often carried out at a potential more negative than the formal potential of the Mn+/M redox couple, E°′, for a suitable time period (seconds to hours), under controlled and known mass transfer conditions. This is the pre-concentration step.| Mn+(aq) + ne− → M(s) | (1) |
| M(s) → Mn+(aq) + ne− | (2) |
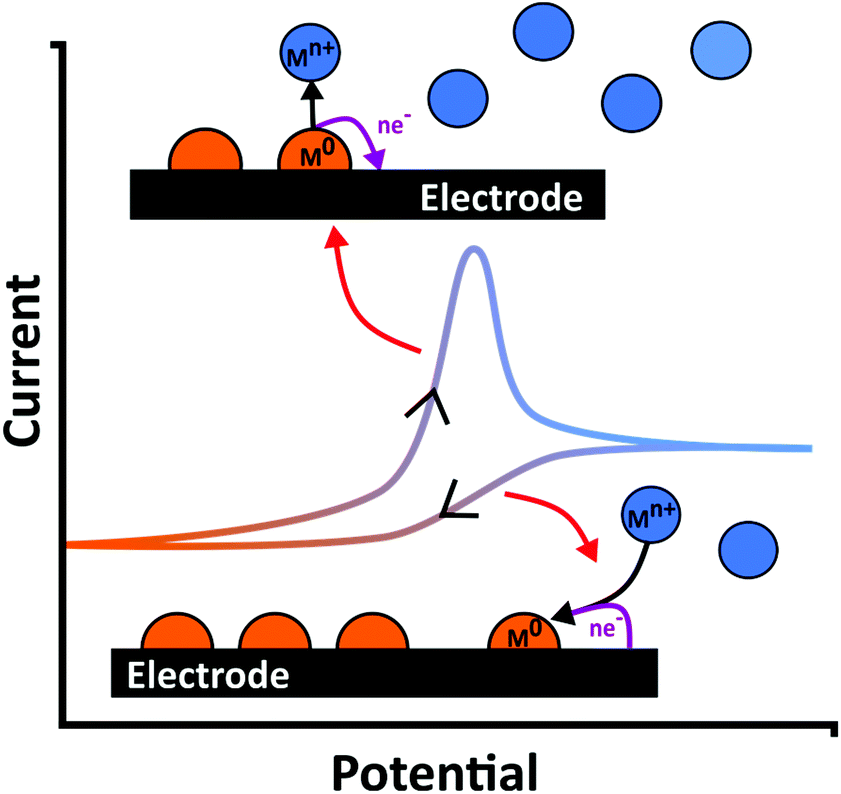 | ||
| Fig. 1 Schematic of metal (M0) deposition and stripping occurring during an exemplar current–voltage curve in a metal ion (Mn+) containing solution. | ||
To improve the LOD the pre-concentration time is typically extended or the rate of deposition increased; the latter achieved by increasing the mass transfer rate at which species can reach the surface.21,31 Pre-concentration is followed by anodic oxidation (or dissolution), of the metal back to metal ions (eqn (2); Fig. 1); the stripping step. Hence electrode material, deposition potential (Edep) and mass transfer are all important factors for consideration (sections 3 and 4).45
Experimentally, analysis of oxidative stripping peaks in the current–potential trace is used to determine metal concentration. The simplest method of performing the stripping step in ASV is by linearly sweeping the current in the anodic direction, although to help further increase detection sensitivity, more complex potential waveforms can be adopted (section 4.3).37 The potential at which the stripping peak occurs and the area under the peak (for a standard linear sweep, this equates to charge42,46) depend on the chemical identity of the species and amount of metal deposited, respectively. In particular, the stripping peak potential can be thermodynamically related to a specific metal through consideration of E°′, for the specific Mn+/M redox couple,47 although this can be more challenging for non-Hg based electrodes (section 3.2). Experimentally peak current/area is related to the original concentration of the species in solution by means of a calibration plot.
Calibrations are determined either by plotting peak height (current) or peak area (charge) vs. concentration. The calibration mode chosen depends on the application. There are exceptions, and some literature use both.48–50 If background processes are present, peaks require background subtraction or baselining.51 Commonly peak height is employed, partially from the historical use of Hg electrodes as many of the theoretical descriptions derived for Hg electrodes, where the peaks are usually well-defined and symmetrical,46 relate concentration to peak current. However, in situations where more than one metal is present, peak shape varies or overlap occurs, the use of peak area can provide better calibration linearity.42,52,53
Metal–metal interactions can cause a varied stripping response; this is discussed further in section 5.4.1. To calibrate for multi metals it is important to understand the interactions. Most often the test solution e.g. river, lake, tap water, is modified to match the calibration solution e.g. in terms of pH. Calibration of the expected metals can be performed individually and then in different ratios to determine how the stripping peaks are affected.54,55 Alternately standard addition or spiking methods can be employed.52,56 This requires knowledge of what metals are likely to exist in the solution matrix.
3. Electrode material
3.1 Mercury
Hg served as an ideal electrode for ASV, as a result of forming homogenous liquid metal amalgams (mercurous alloys), and displaying well defined stripping peaks. Using Hg, a wide range of metals could be detected,30 limited only by anodic dissolution of Hg; Hg cannot be used to analyse Hg or metals with more positive E°′ than its own, e.g. Ag.57 Whilst recent use in the literature is very limited, due to the environmental concerns of Hg, the electrochemical characteristics of Hg which make it ideal for ASV are briefly discussed in order to understand the favourable attributes which are required for alternative electrode development (section 3.2).Hg has the advantages of relatively low capacitance compared to other metals, minimal non-faradaic contributions (from surface oxidation) and a wide cathodic window, due to both the hydrogen evolution reaction (HER; eqn (3)),47 and solvent electrolysis (eqn (4))58 being kinetically less facile compared to other metal surfaces.
| 2H+(aq) + 2e− → H2(g) | (3) |
| 2H2O(l) + 2e− → H2(g) + 2OH−(aq) | (4) |
Even as an Hg-metal amalgam, electrode characteristics in ASV are typically dominated by Hg, rather than that of the dissolved metals.30,59 Given the well-behaved characteristics of Hg electrodes, theoretical models to predict peak shape, position and height i.e. peak current ip have been published and corroborated experimentally.60–62
The two most popular types of Hg electrodes used in stripping analysis are the hanging Hg drop electrode (HMDE) and the Hg thin film electrode (MTFE).36,46 The HMDE has the advantage it can be easily regenerated by simply growing a new drop of Hg, also negating electrode fouling. MTFEs are typically formed by electrodepositing Hg or co-depositing Hg with the metal(s) of interest onto a carbon substrate. This is a practical way of both avoiding handling liquid Hg and generating very thin films.63 The thin films result in exhaustive depletion and narrow stripping peaks, preferred when multiple metals are present to provide increased resolution.48 The smaller volume of Hg, also results in more concentrated amalgam formation, which whilst increasing detection sensitivity, can also exacerbate intermetallic compound formation which lowers the hydrogen overvoltage.36,64 For a MTFE, the Hg film is typically redeposited each time but can be easily removed by simply wiping the electrode. In general, the MTFE approach is far more portable and robust than the drop version.63
3.2 Solid electrodes
With a need to find an alternative to Hg-based electrodes, and as no other metal exists which is liquid at room temperature, solid electrodes have been sought which possess as many of the favourable attributes of Hg as possible, including; retarded HER, low background currents, reproducible surface and narrow stripping peaks whilst also exhibiting low toxicity. Several recent reviews cover the many different types of solid electrodes and chemically modified solid electrodes that have been applied to heavy metal detection by ASV and related techniques.27,65,66 Biological molecules such as DNA, enzymes and bacteria are also increasingly being used as modifiers due to their high specificity.66,67 Examples of metallic electrodes such as Au,13 Ag,68 Pt,61 Ir,69 Bi25 and the more recent, Te,70 can all be found in the literature. In the case of metal electrodes, the possible formation of alloys between deposit and electrode during the metal deposition process, should also be considered as is the case for Bi, which alloys with a variety of metals including Pb, Cd, Sb, Tl, Ga.25 Surface modified or carbon based electrodes have also become increasingly popular in ASV.27,71–73Metal deposition and stripping are more complicated on a solid electrode for a variety of reasons. Firstly, unlike Hg, as soon as the metal is deposited there is a change in surface properties. The surface characteristics are now the combination of the deposit plus the underlying solid electrode, and potentials of deposition, onset potentials for HER and water reduction will change to reflect this. This effect is often overlooked in the literature, but it will affect how metals deposit and the resulting analysis. Moreover, on a solid surface, it is impossible to provide defect free, pristine surfaces, so deposition is likely to be heterogeneous, with a range of different morphologies present.74 This leads to different energies required to strip the metal (or alloy) from the surface and hence broader stripping peaks and changeable peak positions are often observed.75 For example, nanoparticles (NPs) of different sizes, but the same composition, have been observed to strip at different potentials.76,77 It is thus important that the deposition parameters, preparation of the electrode surface etc. are optimised as much as possible to try and move towards a homogeneous, monodisperse distribution of deposited structures on the electrode surface. However, this can be challenging.
Also important for consideration is if too much metal is deposited on the surface it may not be possible to remove all the metal during the stripping step. This becomes an issue when comparing charge passed to predictions from theory,78 and when using the same electrode for repeat deposition/stripping measurements. Incomplete stripping may also arise from conversion of the deposited metal to a less electrochemically active form, such as a metal oxide/hydroxide. This can occur due to reaction with another species, such as oxygen in the solution or electrochemically generated hydroxide ions (OH−).79 Furthermore, when depositing multiple metals, a variety of scenarios could occur; metals may form independently, one metal may plate preferentially on another, intermetallic deposits between the multiple metals could form, or one metal may inhibit or block the deposition of another. In reality several of these could occur at once, again complicating the analysis.
Information on the interaction between a solid electrode and a depositing metal can be inferred by observing the potential at which deposition occurs. The situation is obviously slightly more complicated in the presence of an alloy. For a metal, deposition and stripping theoretically occurs at E°′, but when depositing onto a foreign material, there are three possibilities; under potential deposition (UPD), overpotential deposition (OPD) and alloy formation. UPD occurs when the interaction between the electrode and the metal is stronger, hence more favourable than the metal–metal interaction. A potential lower than E°′ is required for UPD. OPD is where an overpotential, with respect to E°′, is required to induce deposition. The higher the overpotential, the more difficult metal deposition is for that system.80 Theoretical equations do exist for metal stripping on solid electrodes, but there has been limited success, compared to Hg, for fitting theory to experiment. Most papers acknowledge that the complex and stochastic nature of metal deposition on solid electrodes makes prediction both complicated and flawed.81,82 So despite decades of research, no general theory for metal stripping on a solid electrode system has yet been found.
The Bi based electrode is one of the most popular metal electrode materials used in ASV. Bi is typically used in thin film format, the Bi film electrode (BiFE), and is considered to be very similar in electrode characteristics to Hg, but importantly with lower toxicity. BiFE's give narrow, well resolved, reproducible stripping peaks, due to the ability of Bi to form solid metal alloys with a range of metals. Similar results have also been observed for Sb films.83 The Bi film is typically generated by electrodepositing or co-depositing Bi with the analyte ions onto a carbon support electrode. The reason BiFEs are preferred over bulk Bi electrodes is most likely due to the film deposition/co-deposition process being an easy, reliable way of obtaining a reproducible surface. Bulk Bi has a slightly lower HER overpotential than the BiFE because of the differences in crystallinity between film and bulk, resulting in a smaller usable potential range.84 Bi has a lower anodic oxidation potential than Hg, however it has still been used to determine concentrations of metals with more positive stripping potentials than Bi itself, such as Cu.85 Although not explained, this could be due to the formation a Bi–Cu intermetallic which strips at a more anodic potential than Bi; intermetallics are discussed further in section 5.4.1. Reproducibility is best when a new film is deposited each time.25
Carbon materials such as glassy carbon, carbon nanotubes, graphite (highly ordered pyrolytic graphite, edge plane pyrolytic grahite, graphene and even pencil lead) and carbon paste are also good electrode materials for ASV.71,73,86,87 Carbon electrodes are more inert than metals and have low background currents, allowing them to also achieve lower LODs.36,88 They are non-toxic so again are attractive for in vivo or general environmental and biological studies. Their surfaces are easy to covalently modify by synthetic chemistry methods, opening up further possible applications.58,89 Modification of carbon electrodes to improve sensitivity or selectivity in ASV is common place.36,90 For example, conducting polymer layers containing surface molecules that chemically complex metal ions have enabled the simultaneous detection of Pb, Cu and Hg in the range 10−7 to ca. 10−10 M.91,92 Useful ASV metal electrodes, such as Bi and Au have also been added to the carbon surface, typically in NP form by either electrodeposition93 or chemical reduction methods.94 The density of NPs is often such that individual diffusion fields to each NP overlap, reducing the spectacular mass transfer enhancements expected from very small NP electrodes (section 4.2). However, the background signals will be reduced as a result of a reduced amount of active metal on the surface leading to improved signal to noise ratios. The use of modified carbon electrodes for ASV applications has been extensively reviewed elsewhere.71,86
One specific type of carbon electrode worth further discussion is boron doped diamond (BDD). Boron doping is required to turn diamond into an electronic conductor. BDD has been used widely in ASV analysis as it offers the widest potential window of any electrode material in aqueous solution due to the kinetics of water oxidation and reduction being very slow. This is thought to be due to a lack of available catalytic sites on the sp3 surface.88,95 It also presents very low background currents and chemical inertness with the added benefits of mechanical strength, high pressure and temperature resistance and reduced fouling by biomatter and bacterial biofilms, when compared against other electrode materials.96–98 Studies have shown that the stripping potentials for the metals Zn, Cd and Pb, Fig. 2, were not significantly shifted on BDD relative to Hg. Both electrodes were capable of metal detection over a concentration range of 3–4 orders of magnitude, with LODs in the low ppb range.72
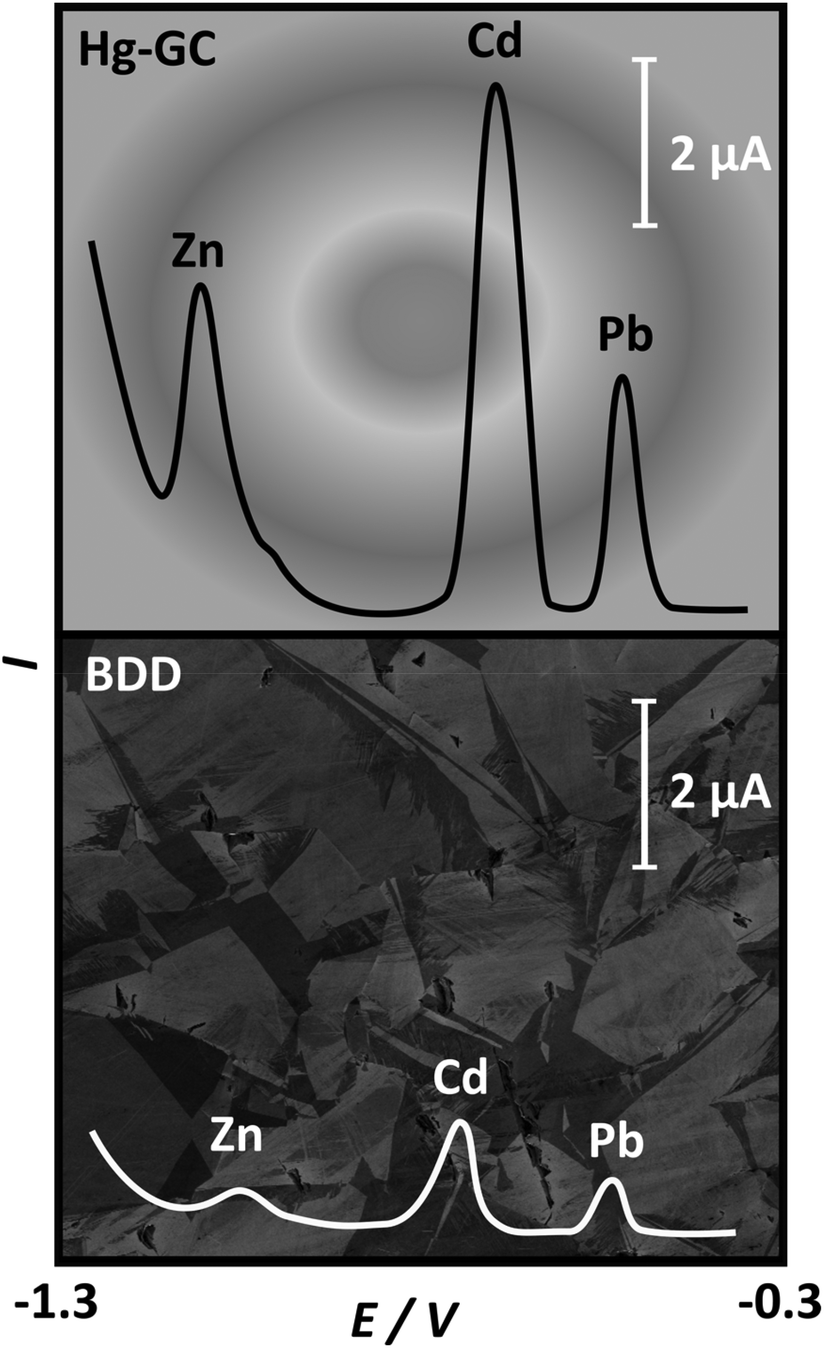 | ||
| Fig. 2 Comparison of stripping voltammetric I–E curves for 100 ppb solutions of Zn, Cd and Pb in acetate buffer, pH 5.2, on a Hg coated glassy carbon electrode (Hg-GC) (top) and BDD (bottom). Adapted from ref. 72 with permission from Elsevier, copyright 2006. | ||
In recent years there has been a greater rise in the use of cheap, disposable and/or multiplexed electrodes for heavy metal detection. The disposable nature, means long term issues with electrode fouling due to placement in the real environment can be negated. However, batch reproducibility does need to be assured in order to have confidence in the results, when multiple electrodes are used for calibration and measurement. The use of multiplexed electrodes on the same platform, means that different electrodes can be employed to extend the range of detectable species.99 Such electrodes are well suited for on-site analysis as they are small and when integrated with appropriate instrumentation, portable. Often, these are screen printed electrodes, where the reference and counter electrodes can be printed alongside the working electrode, producing a compact sensor system. Alternatively, microfabrication procedures can be used to print arrays of metal electrodes.99 Screen printed electrodes can be made from many different materials such as carbon or metal oxide, printed as an ink and integrated into a support. The surface can be further functionalised if required, e.g. through the addition of metal NPs, metal ion complexing groups, depending on the application of interest.100–102 In some cases the support can be made flexible enabling the electrode device to be e.g. worn on the wrist as is the case for the detection of heavy metals in sweat.99
Additionally, the emergence of 3D printing technologies, for the high throughput production of bespoke devices, has also impacted ASV, along with other areas of electrochemical analysis.103 3D printing provides an opportunity to design and print electrodes in non-conventional geometries, and has been used with nanocarbon composite filaments104,105 and metals.106 It has also been used to produce bespoke flow cells to house a more traditional electrode format, e.g. screen printed carbon.103
4. ASV parameters
4.1 Deposition potential
For solutions containing a mixture of metals, in order to ensure deposition of all metals, typically the potential must be made more negative than the most negative E°′ metal. For solid electrodes, it is important to understand the voltammetric behaviour of the metals of interest on the solid electrode of choice before choosing the deposition potential, Edep. Furthermore, the overpotential of deposition (Edep − E°′) is also likely to strongly influence the morphology of the metal deposits formed on the surface. The situation is further complicated when the heterogeneity of the surface is taken into account, be that morphology or surface electroactivity. For example, on BDD which contains heterogeneously doped grains, at moderate overpotentials different morphologies for a single metal can be obtained on differently doped regions of the surface when the grains are significantly large.50 This can result in broadened stripping peaks,50,72,88 compared to e.g. the BiFE or Hg electrode.However, there are limits to the maximum overpotential that can be employed, for example, if Edep is close to the cathodic window, hydrogen gas production (HER) (eqn (3); Fig. 3) results. Bubble formation from HER may block electrode accessibility, changing effective electrode area. Production of OH− due to water or oxygen reduction, will result in local pH rises, which in turn can affect metal speciation or lead to the formation of insoluble metal (hydro)oxides (eqn (5)).107,108
| Mn+(aq) + nOH−(aq) → M(OH)n(s) | (5) |
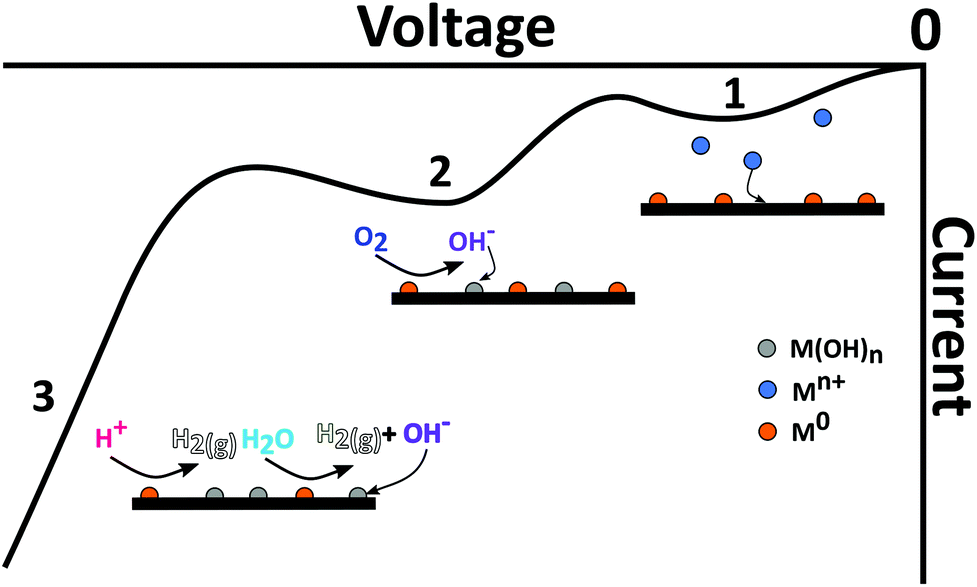 | ||
| Fig. 3 Current–voltage curve in the cathodic direction illustrating the different cathodic processes which could also interfere with (1) metal deposition, and include (2) metal catalysed ORR resulting in transformation of the metal to metal hydroxide and (3) HER from proton and/or water reduction. | ||
If oxygen is present in solution, the oxygen reduction reaction (ORR), via a two or four electron route, is also possible79 (eqn (6) and (7), written for the four electron pathway), catalysed via the metal electrodeposits which are invariably more active than the support electrode. Whether ORR goes via a two or four electron pathway,79 ORR also results in either proton depletion or hydroxide ion formation (pH dependent). Given it occurs at potentials less negative than those described by eqn (3) and (4), ORR is definitely a cause for concern. eqn (3)–(7) are illustrated schematically in Fig. 3.
| O2(aq) + 4H+(aq) + 4e− → 2H2O(l) (acid) | (6) |
| O2(aq) + 2H2O(l) + 4e− → 4OH−(aq) (alkali) | (7) |
Metal hydroxide/oxide formation is a real problem for ASV, as it means the electrodeposited species are no longer in the metallic form and, even though some may remain electrochemically active, the characteristics have changed drastically. This can result in less metal being detected via the M/Mn+ stripping response, leading to an underestimation of the concentration of heavy metal in the original solution.
4.2 Mass transfer considerations
For quantitative analysis it is important that during the metal deposition step, mass transfer of metal ions to the electrode is reproducible and high. The former so the calibration is quantitative, the latter in an effort to reduce analysis times. In lab-based systems, experiments are typically carried out in the presence of excess supporting ions, or buffered solutions, which act to exclude migration from theoretical treatment of the system and negate issues arising from local pH changes due to eqn (3), (4), (6) and (7). To illustrate the enhancements achieved with different mass transport systems we first consider a macroelectrode under diffusion-controlled stationary conditions. The peak current for electrolysis of a redox couple under planar diffusion-controlled conditions, Fig. 4a, is described by the Randles–Sevcik equation (eqn (8)):80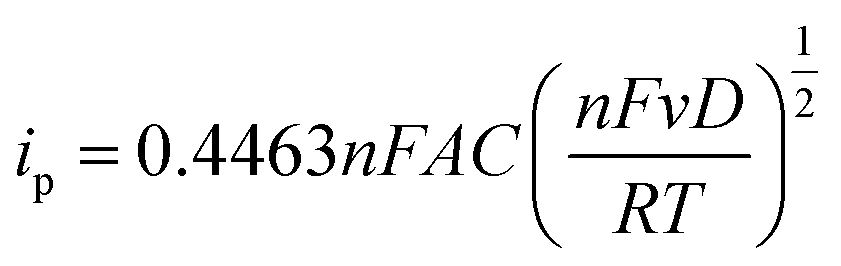 | (8) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000n1/2D1/2v1/2F−1 (for T = 298 K) and for the exemplary case of a 1 mm planar disk electrode (assuming n = 1, D = 1 × 10−5 cm2 s−1 and v = 0.1 V s−1), is 0.0028 cm s−1.
000n1/2D1/2v1/2F−1 (for T = 298 K) and for the exemplary case of a 1 mm planar disk electrode (assuming n = 1, D = 1 × 10−5 cm2 s−1 and v = 0.1 V s−1), is 0.0028 cm s−1.
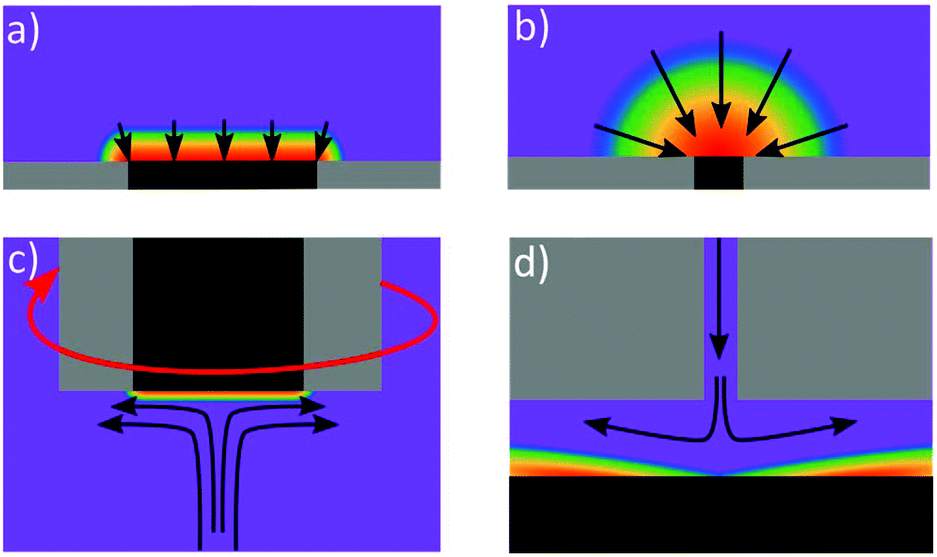 | ||
| Fig. 4 Concentration diffusional profiles at (a) macro electrode, (b) micro electrode (c) rotating disc electrode and (d) wall-jet electrode (adapted from ref. 109 with permission from American Chemical Society, copyright 2010). The arrows represent the flux of electroactive reactant whilst the purple represents the bulk concentration of species, with blue, green, yellow, orange and red representing decreasing concentration. | ||
One way of increasing mass transfer elegantly is by decreasing the size of the electrode such that the diffusional flux profile changes from linear to hemispherical (Fig. 4b),110 such electrodes are referred to as micro or nanoelectrodes. Eqn (9) describes the theoretical steady-state current at an inlaid planar disk micro (and smaller) electrode in stationary solution:
| ilim = 4nrFDc | (9) |
Many ASV systems couple convective flow to diffusion as a means of increasing mass transfer in the system (referred to as hydrodynamics). A popular choice is the rotating disk electrode (RDE)33,46 which provides well defined hydrodynamics and results in the concentration gradient being confined to a much smaller distance from the electrode surface, compared to the stationary macrodisk electrode, Fig. 4c. MTFEs have been used in the RDE format along with Ag,68 Au114 and BDD electrodes,115 in order to increase the rate of metal deposition during the pre-concentration step. The flow hydrodynamics for an RDE are very well understood, being first characterised by Levich and Landau in 1942.116 The Levich equation (eqn (10)) describes the relationship between deposition current (ilim) and rotation frequency, ω (Hz);
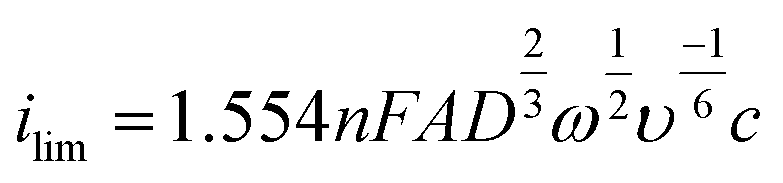 | (10) |
Stripping has been reported under both hydrodynamic and quiescent (diffusion-only) conditions. The most popular method seems to be stripping in quiescent solution, with one argument raised, that forced convection adds to the noise of the system making quantification difficult.123 However, some reports claim stripping should be performed with forced convection as it increases stripping currents.60 If the former is implemented and hydrodynamics have been used to increase mass transfer, a rest period is often implemented before stripping to allow solution to come to rest.53,68,82 Finally, one question that is rarely discussed in the literature, is what is the stability of the metal deposit on the electrode surface under forced convection? Under these conditions metal nanostructures that poorly adhere to the surface, may get removed from the surface during electrodeposition, and are therefore unaccounted for.
4.3 Potential wave forms
One of the simplest voltammetric experiments is linear stripping voltammetry (LSV), where the potential is swept (more accurately “stepped” with digital potentiostats) from one voltage to another at a specified rate (scan rate; Fig. 5a and b). Whilst this is useful for assessing the voltammetric behaviour of metal deposition and stripping on an electrode surface, it contains both the faradaic (from metal stripping) and non-faradaic (background currents such as capacitance) components of the current in the voltammetric window.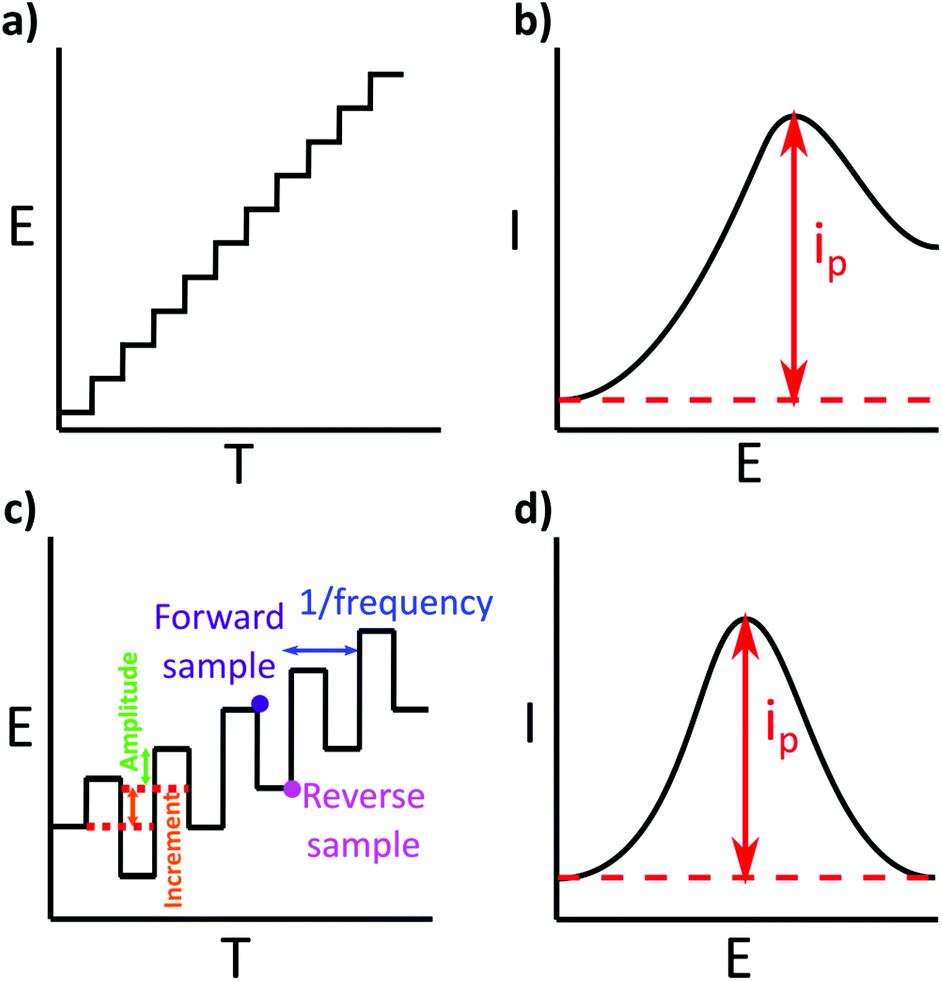 | ||
| Fig. 5 (a) The change in potential over time for a digital LSV with an exemplar current peak shown in b. (c) Is the wave form of SWV and an exemplar current peak shown in d. The peak current in d is typically larger than that of c. | ||
To increase sensitivity other wave forms, which use a series of potential pulses e.g. as shown in Fig. 5c, to generate current–time decay curves per pulse, are typically adopted in ASV analysis. Ideally, the current is sampled at a time in each pulse when the non-faradaic currents have decayed to zero i.e. the remaining current is faradaic only. Text books will say the current is sampled at the last point in the current–time decay curve, however commercial potentiostats will sample over a manufacturer-defined percentage of the pulse.124 The resulting square wave voltammetry (SWV) current should therefore be free of background contributions from non-faradaic currents, enhancing the signal of interest and improving LODs. One example of a voltammetric technique is SWV (Fig. 5c and d), first introduced by Barker in 195237 and later made popular through the work of the Osteryoungs.38,125,126 Others include Normal Pulse (NPV) and Differential Pulse (DPV). DPV and SWV are the most popular, with SWV typically taking less time to run than DPV, due to the use of shorter current pulses.125
Voltammetry is not the only electrochemical method used for heavy metal detection, other potentiostatic, galvanostatic and impedance based methods have been successfully implemented, but are not discussed within the scope of this article.66
5. Experimental practicalities
The range of real world applications is significant with researchers implementing ASV in a wide variety of different environments. Aqueous media such as tap water, waste water, and natural freshwaters are often adjusted prior to measurement, by the addition of buffers, acid or salt.12,17,56,121,127,128 Urine and sweat have been analysed without sample modification e.g. to enable real time measurements as in the case of sweat.99 Seawater has been analysed both as is and with acidification.102 More complex samples such as milk, honey, gasoline, hair, blood, food, soil and sediment require more involved treatment prior to solution analysis e.g. acid digestion, ashing, hydrogen peroxide, red blood cell lysis, and microwave digestion.13–15,17,127,129–131,1325.1 Sample collection and contamination
If an analytical environmental measurement is not made directly at the source, which is very often the case, collection and storage of a sample can impact on the validity of the metal concentration measurement. Ideally samples are collected with as little disruption as possible and with consideration of potential contaminants e.g. oil from a boat engine. The choice of sample container can be a potentially large source of error. Adsorption of metal ions from solution onto, and leaching of trace metals from the container walls are two issues to be aware of, illustrated in Fig. 6a. Metal containing species are sometimes added as catalysts during the polymer container manufacturing process. Any metal impurities present in the walls can be leached when using dilute HNO3 and or HCl solutions. However, solution acidification can also reduce losses from metal adsorption onto container walls.23,133 | ||
| Fig. 6 (a) Composition of a water sample with representative ideal stripping profiles for two metals (green and purple), central image. Wall adsorption can act to reduce the bulk concentration, here purple metal adsorbs more than the green, left image, whilst contamination (right), acts to provide an additional metal (red) and red stripping peak. (b) The composition of a natural water sample, by using a 0.45 μm filter, some components such as bacteria algae and larger silt particles are removed, but others pass through e.g. humic and fulvic acids, clay particles and viruses. (c) Acidification of the filtered sample can result in the desorption of metal ions from humic and fulvic acids. At low pHs humic acids may precipitate out of solution. | ||
Unfortunately, there is no exact information on how, or even whether it is necessary to clean storage bottles before use for trace metal analysis, unless the laboratory is accredited and following standard operating procedures e.g. BS EN ISO 5667. Thus many people adopt a variety of different approaches. Some use time consuming and intensive cleaning procedures,134–136 whilst others report off-the-shelf clean high density polyethylene (HDPE) bottles are satisfactory for (ICP-MS) trace metal analysis,23 even though white HDPE bottles have been shown to be contaminated with high levels of Ba and Zn.137 This could be the reason why contrasting data has been presented. For example, for a PE sample container, but with no information provided on cleaning, one laboratory observed a decrease in metal ion concentration, from a river water sample, over a 10 day storage period,138 whereas, a contrasting study reported no changes in metal concentration, of a freshwater sample, stored for 26 days.20
Ideally, sample collection and storage would be avoided and analysis at the source is ultimately the best solution. If sample collection and storage is required, the researcher is encouraged to consider that metal loss (from solution to container walls) or contamination (from the container wall to solution) are possibilities. An acid clean (e.g. 0.1 M HNO3 overnight, and rinsed thoroughly) could be employed but only if shown to reduce, and not cause contamination.
5.2 Speciation, adsorption and pH
In natural environments heavy metals are found in a variety of forms. Whilst the simplest is the free hydrated metal ion, metals can also form complexes with organic and inorganic molecules, adsorb to colloidal organic and inorganic compounds, as well as being found in particulate mineral forms. Inorganic anions like Cl−, SO42− and HCO3− and organic compounds such as humic and fulvic acids will also complex with metal ions.139 Al, Si, Mn and Fe oxides and clay minerals adsorb metals to their surface, Fig. 6b. Adsorbed metal–colloidal particles themselves have an environment dependent stability;139 see Pourbaix diagrams for potential/pH dependence. The same is true also of biological environments. For example, in the case of blood, heavy metals such as Pb are found mainly bound to haemoglobin.132As the pH of the system decreases, metal-ligand speciation changes. For example, metal hydroxide species can exist as hydrated metal ions and adsorbed metal cations are released into solution. The overall result is an increase in the number of free metal ions in solution as pH decreases, which directly translates to increasing stripping peak currents, Fig. 7. Hence knowledge of pH dependant metal-ligand speciation curves and Pourbaix diagrams is very useful. Thus, for ASV in real samples it is very important to recognise the role pH plays in the results observed. This concept has been illustrated experimentally using a dual electrode arrangement consisting of a closely placed pH generating electrode and ASV detector electrode in a ring-disk geometry. Via the generation of protons through electrolysis of water at the ring, it was possible to quantifiably decrease the pH of the disk electrode measurement environment.140,141 For both Hg and Cu detection on BDD ring-disk electrodes, the stripping currents were shown experimentally to increase as the local pH decreased.140,141
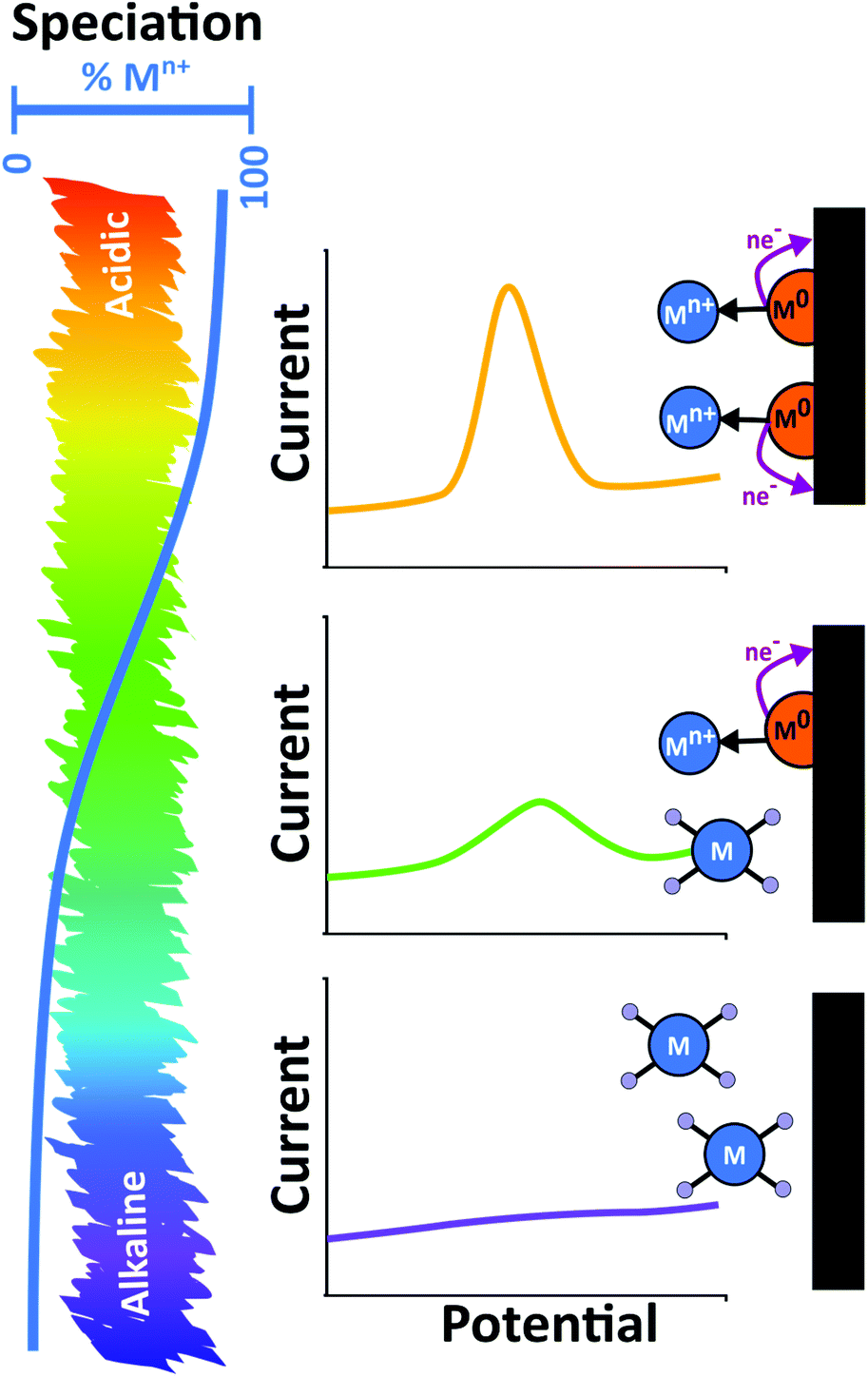 | ||
| Fig. 7 The effect of solution pH on speciation of metal ions and the resulting stripping behaviours. As pH increases a reduction of the stripping peak is observed as fewer metal ions are deposited in the metallic state. | ||
ICP methods, as they generally use acidified solutions, provide a “total” metal concentration in the sample but no information about speciation, and therefore how much of the metal concentration may pose a toxicity risk. If speciation is required, ICP-MS must be coupled with alternative analytical techniques, e.g. capillary electrophoresis, liquid, gas or ion chromatography.142–147 In contrast, ASV can provide information on toxicity levels. For example, ASV was found to produce Cu concentration data that correlated well with its associated toxic fraction, e.g. for seawater spiked with Cu.148 In an interesting variant of ASV, both fast scan deposition149 and adsorption149 voltammetry have been used to provide real time environmental information on metal ion speciation (via rapid measurement of the free metal concentration).
Often samples are filtered (typically with a 0.45 μm pore size filter) to remove any larger insoluble particulates. This process also removes any metal ions bound to the surface of these particulates. The filtered solution is considered the dissolved fraction but will still include smaller insoluble colloids, along with humic and fulvic acids, Fig. 6b. Acidification of the filtered sample typically results in desorption of metal ions from these species, Fig. 6c. Note, rapid acidification of the solution can also cause humic acids to become insoluble, trapping metal ions before they have a chance to desorb.139
For some metals, identification of the original redox state is important as this can drastically affect toxicity, adsorption and mobility of the metal. Electrochemical methods can be used to distinguish between valence states. For example, CrIII is considered an essential trace element, required in the glucose tolerance factor essential for normal glucose metabolism.150 In contrast, CrVI is toxic as it is an oxidising agent and forms free radicals during reduction to CrIII inside cells. By careful choice of deposition potential, it is possible to selectively deposit CrVI over CrIII, as has been demonstrated for artificial seawater.128
Arsenic is generally found as inorganic AsIII and AsV, both of which are highly toxic.151 AsIII is soluble and can be reduced to As0. As0 has low solubility in Hg, but this has been improved by the addition of selenite or Cu which cause the formation of Hg soluble compounds.32 Au has shown the most promise for AsIII detection either as the electrode material or as a surface modification, e.g. in the form of nanoparticles. On Au, detection is possible over a wide pH range as Au has a high overpotential for hydrogen, allowing As to be deposited without HER.32,152 Electrodes such as Pt are more affected by HER. Au also shows higher and sharper As stripping peaks than Pt.153 In contrast, AsV requires an extreme negative potential, making it unfeasible to deposit due to the extent of HER that occurs. Therefore a chemical reduction to AsIII is first required before an electrochemical reduction step to give total As.153,154
For many ASV measurements reported in the literature some form of pH adjustment is often included as part of the solution preparation process. pH adjustments are typically made by addition of an acid (e.g. HNO3, HCl) or buffer.21,31,118 As discussed, it is important to then reflect how closely the ASV measurement of the free/labile metal ions represents the original solution at the source. Local pH increases at the electrode surface during electrodeposition, due to eqn (3), (4), (6) and (7), should not be neglected and is one reason why buffers are added. Acetate (pH ≈ 4–5.6) buffers are most commonly used for ASV,15,17,20,31,36,121,128,155 although phosphate156 (pH ≈ 7) and citrate21 (pH ≈ 6) have also been employed. Buffer ions should weakly bind to the metal ions to ensure they remain labile in solution.157,158 If speciation is an important consideration minimal change to the sample solution is preferred. If some modification is required, it is useful to match the pH of the test solution as closely as possible to that of the natural environment.
Many environmental samples, are naturally buffered with carbonate,159 although ammonia, silicate, borate and phosphate,160 can also play a small buffering role.161 Environmental pH typically lies in the range 6–9. The buffer capacity will vary depending on the source, for example, seawater has a higher buffer capacity than freshwater due to the increased salt content. In aquatic water systems, during periods of high photosynthetic activity, removal of CO2 from waters can result in an increase in the pH of the system. Conversely, acidic inputs in the form of acid mine drainage or acid rain can also lower the pH.162
5.3 Oxygen
For the reasons discussed in section 4.1, oxygen presence is problematic due to ORR causing a local pH increase due to proton depletion (eqn (6)) or hydroxide production (eqn (7)),163 and metal hydro(oxide) formation.164 The easiest way to combat ORR issues is to deoxygenate, although buffering the solution can negate electrochemically induced pH changes. Alternatively, the use of strongly acidic solutions makes local changes in pH less apparent. The deoxygenation of real samples however can cause CO2 removal and as a result increase the solution pH which, depending on the pH change, may also affect the speciation.1655.4 Interferences
Intermetallic compounds formed on electrode surfaces can strip very differently to pure metal deposits, and therefore cannot be simply treated as a sum of their constituent metals. Intermetallics can form as a result of alloying with the electrode e.g. during co-deposition with Bi, or with other metals present in the solution, during the deposition process. For Hg electrodes, the latter is most important and intermetallic interferences for Cu–Zn, Zn–Cd,33,64 Cu–Ni and Cu–Cd,80 especially at high metal concentrations (small Hg volumes), were found to be problematic. For example, formation of intermetallic Cu–Zn resulted in the depression of the Zn peak, and an “apparent” enhancement of the Cu peak. However, the latter was due to Cu–Zn stripping at a very similar potential to that of Cu, so much so that the two peaks could not be resolved.64 Cu–Zn formation can be prevented by using Ga which preferentially forms a Ga–Cu intermetallic compound. This method of adding a “third element” was successful for several other intermetallic interferences on MFEs.170
On solid electrodes intermetallic compounds are also problematic,85 more so than for Hg, as pre-concentration occurs only on the surface where interactions between different metals, are very likely, except at very low concentrations. Further examples of troublesome intermetallic compounds at solid electrodes include, Cu–Zn,171 Cd–Pb,172 Ni–Cu–Zn173,174 and Fe with Cu, Pb, Cd and Zn.158
The stripping potentials for two metals that have deposited independently (not as an intermetallic compound) can also sometimes overlap (e.g. Cd/Tl), making quantification difficult.36,175 However, there are several combinations of multi-metals that can be simultaneously detected without issue, but this depends on factors such as electrode material, relative concentrations of the metals, deposit morphologies, etc.119,176 This is illustrated by Fig. 8 where Bi can both act in an analogous way to Hg for some metals, and form binary intermetallic complexes with others. In the example given Bi forms an intermetallic compound with Cd, making its stripping potential more negative and enabling resolution from the Tl peak.85,177
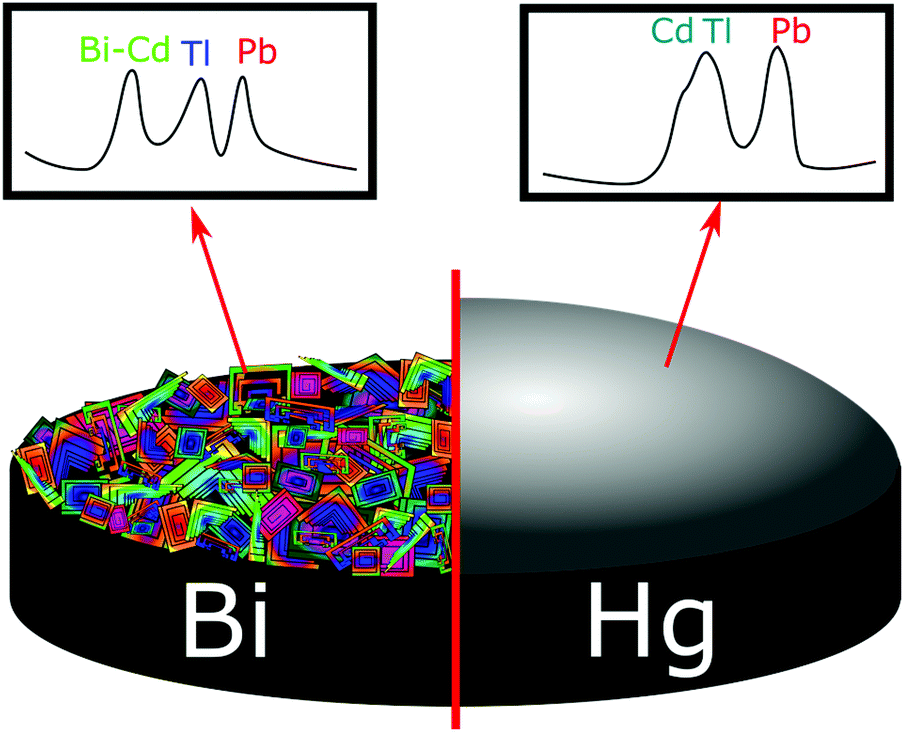 | ||
| Fig. 8 Differences in stripping voltammograms of a mixture of 50 μg L−1 Pb2+, Cd2+, and Tl+ on Bi and Hg thin-film electrodes. Solutions, 0.1 M acetate buffer (pH 4.5) containing 400 μg L−1 Bi or 10 mg L−1 Hg. Deposition for 120 s at −1.2 V. Adapted from ref. 175 with permission from American Chemical Society, copyright 2000. | ||
Interfering metals can also be removed prior to ASV analysis using pre-treatment processes or methods such as ion exchange resins.178 The interference of Cu–Cd and Cu–Pb intermetallics can be mitigated by the addition of ferrocyanide to form an inert Cu complex in solution, but renders the Cu unquantifiable.172 Ion exchange resins have been used to both remove interfering Mn, Fe, Cu and Ni from solution, prior to the detection of inorganic As179 and determine labilities of soluble metals.180
Finally, detecting fewer metals at a time can help reduce possible metal–metal interference effects. This can be achieved using e.g. a chemically modified electrode that selectively accumulates specific metals due to strong interactions of the metal of interest with specific groups on the modifying material leading to improved resolution,127 or by varying the deposition potential to plate only metals with the more positive E°′s.173
Organic matter e.g. proteins, fats, surfactants and bacterial films, can also react with or block the surface of the electrode173,185 affecting stripping signals.186 Polishing the electrode in between measurements187 or UV treatment, can be used to remove or prevent these films (section 5.7). There are methods postulated to combat most common interferences, but some trial and error is still likely when establishing a method.
5.5 Conductivity
The addition of a supporting electrolyte or buffer is common in electrochemical studies, in order to negate migration, simplifying analysis of the resulting current to diffusion or diffusion-convective considerations. It also helps to minimise Ohmic drop due to the lowering of the solution resistance. If measuring low conductivity solutions, such as tap water, lake or river waters (100–2000 μS cm−1), adding a supporting electrolyte or buffer is helpful but any change in solution pH must be considered. If the solution is left in its original state it is important to recognise Ohmic drop will result in a negative shift in potential for effective deposition.188 This is not an issue, however, in seawater due to the high salt concentrations (20000–50000 μS cm−1).189,190 In low conductivity solutions, one option to negate Ohmic drop is to use microelectrodes (section 4.2).188
5.6 Temperature
Temperature affects kinematic viscosity, diffusion coefficient, equilibrium potentials, the extent of reactions such as gas evolution and interactions between species in solution. These result in a complex series of changes to a system as temperature is varied.191 With respect to ASV analysis, temperature has been found to cause shifts in peak potentials, changes in peak current192 and can cause degradation of some modified electrodes, such as EDTA bonded conducting polymer modified electrodes, where a drop in signal was observed as the temperature was increased from 30–50 °C.92During electrodeposition, temperature variations are problematic for the above reasons, as such temperature is an important parameter to keep constant during deposition and stripping; most measurements in the laboratory take place under ambient conditions. Temperature changes can be accommodated, however they must be known and controlled in order to bring advantages. For example, induced temperature gradients in solution can be used to generate convective flow, which both increases mass transfer during the deposition step and the electron transfer kinetics,193,194 resulting in enhanced stripping currents compared to room temperature measurements. This is particularly effective when the induced temperature changes are very localised and controlled, unlike water bath heating or hotplate heating of the electrode.195 Joule heating,196 laser heating164 and microwave heating197 (both pulsed and continuous194) have all been employed to increase mass transfer in a controlled manner during the metal deposition step.
Note, natural variations in temperature pose a challenge to electrochemical sensing in the real environment. Integrated temperature measurements are one way to account for variation. Further, in the research laboratory, measurements often negate testing the device at the environmentally relevant temperature.
5.7 Ultra Violet (UV) irradiation
UV irradiation destroys organic matter such as humic and amino acids, causing the release of bound metal ions, and also preventing organic processes from changing the sample composition after collection.178 Metal bound-inorganic complexes tend to be unaffected by the irradiation process and thus the free, organic bound and total metal concentrations can be deduced using a combination of UV and acid digestions.21,31 The presence of organic compounds is known to interfere greatly with metal concentration analysis, through either biofouling of the electrode surface or interactions with the metals of interest. For example, As is particularly affected by the presence of organic matter by sorption, thus samples are acidified and irradiated, then subjected to a further acid digestion to obtain total concentration.32,198 UV has the advantage of lowering the risk of contamination as no reagents are being added. There are reports of issues with irradiation of freshwaters where it is thought to cause losses due to re-adsorption of the metals released by organic matter decomposition by precipitated iron oxide particles.199,2005.8 Electrode cleaning
The condition of the electrode surface is very important in electrodeposition. Any residual deposit can alter the electrode performance. Returning the electrode to its initial condition after the stripping step has occurred can require a cleaning step in the procedure, either mechanically or electrochemically. Mechanical cleaning involves physically polishing the electrode using some form of polishing pad typically impregnated with alumina particles.201,202 This is not practical for either an in situ sensor that would be left in the environment for prolonged periods of time or for many of the disposable type sensors in use today, due to perturbation of the electrode structure. Electrochemical cleaning can be effective in renewing the electrode surface, especially if the metal stripping step is incomplete and metal deposits still remain on the surface. An electrochemical clean generally involves cycling or holding the electrode at a predetermined oxidative potential for a defined period of time, oxidising the metal and forming a locally acidic environment to aid dissolution. This has been used successfully to remove deposited metals from BDD50 and Bi electrodes.25If the measurement is made in the real solution the electrode can foul due to adsorption of contaminants or bacteria onto the surface. Membranes can be added to reduce this effect, for example, Nafion has been used to protect the surface of BiFE's from fouling by large macromolecules including Triton X-100, gelatin,203 albumin, humic acid204 and proteins.99 Bacterial attachment is especially problematic as bacteria proliferate to form biofilms. Whilst the choice of electrode material can reduce the rate of biofilm formation,97,98 many of the current commercial sensors e.g. pH, dissolved oxygen and conductivity, aimed at long term use in water environments use mechanical brush wipers to remove biofilm from the sensor surface.205
6. Summary of considerations
Successfully implementing ASV can be challenging, below we highlight the main issues for consideration.Before starting analysis, it is useful to consider the metals to detect, the required LOD and what is known about the sample solution, be it river, lake, sea, tap water etc. The latter considers parameters such as, other metals or interferences, particulates, pH of solution, buffer capacity and conductivity. The answers to these questions will then help inform on the set-up, which includes the electrode material, geometry and functionalisation (if appropriate), the method of mass transfer enhancement to reach the required LOD, relevant deposition and stripping parameters, whether intermetallic formation is expected, and methods to mitigate possible metal interference effects.
Also important is whether the original solution taken from the source has been altered in any way for analysis. Most likely this will involve a pH adjustment. If so, the impact on the speciation of metal ions in solution must be considered. Making the solution acidic will push all metals into an electrochemically detectable form, but this does not necessarily reflect the toxicity/bioavailability of the original system. To negate local pH changes during the electrochemical measurement, due to e.g. ORR, addition of a buffer is extremely useful, but consideration again must be made to the difference in pH between the measurement system and the real source. For low conductivity solutions, such as tap and river water, in order to negate migration, addition of excess ions in buffer or supporting electrolyte form is often necessary.
Calibration of the electrode is very important, and this is often made in model solutions containing different concentrations of the metal of interest under the same conditions the test solution will be run e.g. of pH, buffer or added electrolyte conditions. Alternatively spiking of the test solution with known, added concentrations of the metal(s) of interest has also been adopted. Cleaning of the electrode in between metal depositions is also important in order to ensure no residual metal is left over on the electrode surface for the next measurement. If disposable electrodes are to be used, then the reproducibility of the electrode between different electrodes from the same batch is an important consideration.
Often neglected is the container used for solution collection and assessment of the effectiveness and impact of the vessel cleaning procedure on the measurement. Comment should also be made on whether the sample solution has been filtered, acidified or UV irradiated prior to measurement, as these can all impact measurements e.g. removing larger particles onto which metal ions can be adsorbed, causing precipitation, releasing metal ions from organic matter, changing speciation.
In summary, ASV has the potential to be a very powerful technique but does require optimisation for each application in order to be effective. This is perhaps the reason for limited commercial activity despite impressive achievements by ASV in the scientific literature.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Centres for Doctoral Training in Diamond Science and Technology, with Bruker (EP/L015315/1) and CENTA (NE/L002493/1) for funding, for AJB and NER, respectively. Thanks also to Dr Haytham Hussein, Dr Tania Read, Samuel Cobb, Lee Simcox and the Warwick Electrochemistry Group for useful discussions and advice. JVM would like to thank the Royal Society for an Industry Fellowship (INF/R1/180026).Notes and references
- P. B. Tchounwou, C. G. Yedjou, A. K. Patlolla and D. J. Sutton, Mol. Clin. Environ. Toxicol., 2012, 101, 133–164 CrossRef PubMed.
- Council Directive 2008/105/EC on environmental quality standards in the field of water policy, Off. J. Eur. Union, 2008, L348/84, 84–97.
- M. Olivares and R. Uauy, Am. J. Clin. Nutr., 1996, 63, 791–796 CrossRef PubMed.
- H. A. Schroeder, A. P. Nason, I. H. Tipton and J. J. Balassa, J. Chronic Dis., 1967, 20, 179–210 CrossRef CAS PubMed.
- H. Needleman, Annu. Rev. Med., 2004, 55, 209–222 CrossRef CAS PubMed.
- A. Martelli, E. Rousselet, C. Dycke, A. Bouron and J. M. Moulis, Biochimie, 2006, 88, 1807–1814 CrossRef CAS PubMed.
- J. Godt, F. Scheidig, C. Grosse-Siestrup, V. Esche, P. Brandenburg, A. Reich and D. A. Groneberg, J. Occup. Med. Toxicol., 2006, 1, 1–6 CrossRef.
- P. M. Chapmann, H. E. Allen, K. Godtfredsen and M. N. Z'Graggen, Environ. Sci. Technol., 1996, 30, 448A–452A CrossRef.
- M. Kazemipour, M. Ansari, A. Mohammadi, H. Beitollahi and R. Ahmadi, J. Anal. Chem., 2009, 64, 65–70 CrossRef CAS.
- S. M. Rosolina, J. Q. Chambers, C. W. Lee and Z. L. Xue, Anal. Chim. Acta, 2015, 893, 25–33 CrossRef CAS PubMed.
- S. M. Rosolina, J. Q. Chambers and Z. L. Xue, Anal. Chim. Acta, 2016, 914, 47–52 CrossRef CAS PubMed.
- G. Hughes, K. Westmacott, K. C. Honeychurch, A. Crew, R. M. Pemberton and J. P. Hart, Biosensors, 2016, 6, 50–89 CrossRef PubMed.
- A. Giacomino, A. Ruo Redda, S. Squadrone, M. Rizzi, M. C. Abete, C. La Gioia, R. Toniolo, O. Abollino and M. Malandrino, Food Chem., 2017, 221, 737–745 CrossRef CAS PubMed.
- A. Sani and I. L. Abdullahi, Toxicol. Rep., 2017, 4, 72–76 CrossRef CAS PubMed.
- A. K. Jaiswal, S. Das, V. Kumar, M. Gupta and N. Singh, Int. J. Engine Res., 2015, 4, 235–239 CrossRef CAS.
- G. Liang, L. Pan and X. Liu, Int. J. Environ. Res. Public Health, 2017, 14, 1–10 Search PubMed.
- G. Kefala, A. Economou, A. Voulgaropoulos and M. Sofoniou, Talanta, 2003, 61, 603–610 CrossRef CAS PubMed.
- G. E. Batley and T. M. Florence, Anal. Lett., 1976, 9, 379–388 CrossRef CAS.
- T. M. Florence, G. E. Batley and P. Benes, CRC Crit. Rev. Anal. Chem., 1980, 9, 219–296 CAS.
- T. M. Florence, Water Res., 1977, 11, 681–687 CrossRef CAS.
- T. M. Florence, Talanta, 1982, 29, 345–364 CrossRef CAS PubMed.
- J. W. Olesik, Anal. Chem., 1991, 63, 12A–21A CrossRef CAS.
- C. Reimann, U. Siewers, H. Skarphagen and D. Banks, Sci. Total Environ., 1999, 239, 111–130 CrossRef CAS PubMed.
- A. B. M. Helaluddin, R. S. Khalid, M. Alaama and S. A. Abbas, Trop. J. Pharm. Res., 2016, 15, 427–434 CrossRef CAS.
- J. Wang, Electroanalysis, 2005, 17, 1341–1346 CrossRef CAS.
- G. Gillain, G. Duyckaerts and A. Disteche, Anal. Chim. Acta, 1979, 106, 23–37 CrossRef CAS.
- Y. Lu, X. Liang, C. Niyungeko, J. Zhou, J. Xu and G. Tian, Talanta, 2018, 178, 324–338 CrossRef CAS PubMed.
- M. G. Tamba and N. Vantini, J. Electroanal. Chem., 1970, 25, 235–244 CrossRef CAS.
- G. M. S. Alves, L. S. Rocha and H. M. V. M. Soares, Talanta, 2017, 175, 53–68 CrossRef CAS PubMed.
- J. Wang, Stripping analysis : principles, instrumentation, and applications, VCH, Weinheim, 1985 Search PubMed.
- T. M. Florence, Analyst, 1986, 111, 489–505 RSC.
- H. Greschonig and K. J. Irgolic, Appl. Organomet. Chem., 1992, 6, 565–577 CrossRef CAS.
- M. L. Tercier and J. Buffle, Electroanalysis, 1993, 5, 187–200 CrossRef CAS.
- K. Pecková, J. Musilová and J. Barek, Crit. Rev. Anal. Chem., 2009, 39, 148–172 CrossRef.
- P. Manisankar, G. Selvanathan, S. Viswanathan and H. Gurumallesh Prabu, Electroanalysis, 2002, 14, 1722–1727 CrossRef.
- J. Wang, Anal. Electrochem., 2006, 70 Search PubMed.
- G. C. Barker and I. L. Jenkins, Analyst, 1952, 77, 685–696 RSC.
- G. C. Barker and A. W. Gardner, Analyst, 1992, 117, 1811–1828 RSC.
- P. Zuman, Crit. Rev. Anal. Chem., 2001, 31, 281–289 CrossRef CAS.
- B. Elema, Antonie Van Leeuwenhoek, 1947, 12, 243–256 CrossRef CAS PubMed.
- C. Zbinden, Bull. Soc. Chim. Biol., 1931, 13, 35–40 CAS.
- W. D. Ellis, J. Chem. Educ., 1973, 50, A131 CrossRef CAS.
- World Health Organisation, Trace elements in human nutrition and health, 1996 Search PubMed.
- United Nations Environment Programme, Minimata Convention on Mercury, 2017 Search PubMed.
- R. D. DeMars and I. Shain, Anal. Chem., 1957, 29, 1825 CrossRef.
- T. R. Copeland and R. K. Skogerboe, Anal. Chem., 1974, 46, 1257A–1268a CrossRef.
- R. G. Compton and C. E. Banks, Understanding Voltammetry, Imperial College Press, 2010 Search PubMed.
- Z. Stojek and Z. Kublik, J. Electroanal. Chem., 1979, 105, 247–259 CrossRef CAS.
- J. Duay, J. E. Ortiz-Santiago and T. N. Lambert, Electroanalysis, 2017, 29, 2685–2688 CrossRef CAS.
- L. A. Hutton, M. E. Newton, P. R. Unwin and J. V. Macpherson, Anal. Chem., 2011, 83, 735–745 CrossRef CAS PubMed.
- A. J. Bard, Electroanalytical Chemistry, A Series of Advances, M. Dekker, 1966 Search PubMed.
- R. G. Clem, G. Litton and L. D. Ornelas, Anal. Chem., 1973, 45, 1306–1317 CrossRef CAS.
- J. Wang, X. Cai, C. Jonsson and M. Balakrishnan, Electroanalysis, 1996, 8, 20–24 CrossRef CAS.
- A. Manivannan, R. Kawasaki, D. A. Tryk and A. Fujishima, Electrochim. Acta, 2004, 49, 3313–3318 CrossRef CAS.
- D. Zhao, X. Guo, T. Wang, N. Alvarez, V. N. Shanov and W. R. Heineman, Electroanalysis, 2014, 26, 488–496 CrossRef CAS.
- W. Kang, X. Pei, C. A. Rusinek, A. Bange, E. N. Haynes, W. R. Heineman and I. Papautsky, Anal. Chem., 2017, 89, 3345–3352 CrossRef CAS PubMed.
- D. W. M. Arrigan, Analyst, 1994, 11, 1953–1966 RSC.
- F. Scholz, Electroanalytical Methods, 2nd edn, 2010 Search PubMed.
- O. Mikkelsen and K. H. Schroder, Electroanalysis, 2003, 15, 679–687 CrossRef CAS.
- C. M. A. Brett and A. M. Oliveira Brett, J. Electroanal. Chem., 1989, 262, 83–95 CrossRef CAS.
- K. Z. Brainina, Talanta, 1971, 18, 513–539 CrossRef CAS PubMed.
- R. S. Nicholson and I. Shain, Anal. Chem., 1964, 36, 706–723 CrossRef CAS.
- T. M. Florence, J. Electroanal. Chem., 1970, 27, 273–281 CrossRef CAS.
- T. R. Copeland, R. A. Osteryoung and R. K. Skogerboe, Anal. Chem., 1974, 46, 2093–2097 CrossRef CAS.
- M. B. Gumpu, S. Sethuraman, U. M. Krishnan and J. B. B. Rayappan, Sens. Actuators, B, 2015, 213, 515–533 CrossRef CAS.
- B. K. Bansod, T. Kumar, R. Thakur, S. Rana and I. Singh, Biosens. Bioelectron., 2017, 94, 443–455 CrossRef CAS PubMed.
- L. Cui, J. Wu and H. Ju, Biosens. Bioelectron., 2015, 63, 276–286 CrossRef CAS PubMed.
- M. Brand, I. Eshkenazi and E. Kirowa-Eisner, Anal. Chem., 1997, 69, 4660–4664 CrossRef CAS.
- M. A. Nolan and S. P. Kounaves, Anal. Chem., 1999, 71, 3567–3573 CrossRef CAS.
- A. Bobrowski, A. Królicka, J. Śliwa and J. Zarębski, Electrochim. Acta, 2017, 252, 453–460 CrossRef CAS.
- N. Y. Stozhko, N. A. Malakhova, M. V. Fyodorov and K. Z. Brainina, J. Solid State Electrochem., 2008, 12, 1185–1204 CrossRef CAS.
- E. A. McGaw and G. M. Swain, Anal. Chim. Acta, 2006, 575, 180–189 CrossRef CAS PubMed.
- M. Hersey, S. N. Berger, J. Holmes, A. West and P. Hashemi, Anal. Chem., 2019, 91, 27–43 CrossRef CAS PubMed.
- S. E. Ward Jones, F. G. Chevallier, C. A. Paddon and R. G. Compton, Anal. Chem., 2007, 79, 4110–4119 CrossRef CAS PubMed.
- D. Menshykau and R. G. Compton, J. Phys. Chem. C, 2009, 113, 15602–15620 CrossRef CAS.
- O. S. Ivanova and F. P. Zamborini, Anal. Chem., 2010, 82, 5844–5850 CrossRef CAS PubMed.
- D. K. Pattadar, J. N. Sharma, B. P. Mainali and F. P. Zamborini, Curr. Opin. Electrochem., 2019, 13, 147–156 CrossRef CAS.
- M. E. Hyde, C. E. Banks and R. G. Compton, Electroanalysis, 2004, 16, 345–354 CrossRef CAS.
- E. Yeager, Electrochim. Acta, 1984, 29, 1527–1537 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, Wiley, New York, 2 edn, 2000 Search PubMed.
- T. Berzins and P. Delahay, J. Am. Chem. Soc., 1953, 75, 555–559 CrossRef CAS.
- S. P. Perone and H. E. Stapelfeldt, Anal. Chem., 1966, 38, 796–799 CrossRef CAS.
- E. Tesarova, L. Baldrianova, S. B. Hocevar, I. Svancara, K. Vytras and B. Ogorevc, Electrochim. Acta, 2009, 54, 1506–1510 CrossRef CAS.
- R. Pauliukaite, S. B. Hočevar, B. Ogorevc and J. Wang, Electroanalysis, 2004, 16, 719–723 CrossRef CAS.
- J. Wang, J. Lu, Ü. A. Kirgöz, S. B. Hocevar and B. Ogorevc, Anal. Chim. Acta, 2001, 434, 29–34 CrossRef CAS.
- W. Zhang, S. Zhu, R. Luque, S. Han, L. Hu and G. Xu, Chem. Soc. Rev., 2016, 45, 715–752 RSC.
- S. Sharma, J. Electrochem. Soc., 2020, 167, 037501 CrossRef.
- J. V. Macpherson, Phys. Chem. Chem. Phys., 2015, 17, 2935–2949 RSC.
- W. E. Van der Linden and J. W. Dieker, Anal. Chim. Acta, 1980, 119, 1–24 CrossRef CAS.
- D. W. M. Arrigan, Analyst, 1994, 119, 1953–1966 RSC.
- A. Mandil, L. Idrissi and A. Amine, Microchim. Acta, 2010, 170, 299–305 CrossRef CAS.
- M. A. Rahman, M.-S. Won and Y.-B. Shim, Anal. Chem., 2003, 75, 1123–1129 CrossRef CAS PubMed.
- A. Economou, Sensors, 2018, 18, 1–23 CrossRef PubMed.
- L. Cui, J. Wu and H. Ju, Chem. – Eur. J., 2015, 21, 11525–11530 CrossRef CAS PubMed.
- S. Alehashem, F. Chambers, J. W. Strojek, G. M. Swain and R. Ramesham, Anal. Chem., 1995, 67, 2812–2821 CrossRef CAS.
- R. Trouillon and D. O'Hare, Electrochim. Acta, 2010, 55, 6586–6595 CrossRef CAS.
- L. J. Simcox, R. P. A. Pereira, E. M. H. Wellington and J. V. Macpherson, ACS Appl. Mater. Interfaces, 2019, 11, 25024–25033 CrossRef CAS PubMed.
- R. E. Wilson, I. Stoianov and D. O'Hare, Electrochem. Commun., 2016, 71, 79–83 CrossRef CAS.
- W. Gao, H. Y. Y. Nyein, Z. Shahpar, H. M. Fahad, K. Chen, S. Emaminejad, Y. Gao, L. C. Tai, H. Ota, E. Wu, J. Bullock, Y. Zeng, D. H. Lien and A. Javey, ACS Sens., 2016, 1, 866–874 CrossRef CAS.
- A. Hayat and J. L. Marty, Sensors, 2014, 14, 10432–10453 CrossRef CAS.
- M. Li, Y. T. Li, D. W. Li and Y. T. Long, Anal. Chim. Acta, 2012, 734, 31–44 CrossRef CAS PubMed.
- B. Molinero-Abad, D. Izquierdo, L. Pérez, I. Escudero and M. J. Arcos-Martínez, Talanta, 2018, 182, 549–557 CrossRef CAS PubMed.
- Q. Sun, J. Wang, M. Tang, L. Huang, Z. Zhang, C. Liu, X. Lu, K. W. Hunter and G. Chen, Anal. Chem., 2017, 89, 5024–5029 CrossRef CAS PubMed.
- K. C. Honeychurch, Z. Rymansaib and P. Iravani, Sens. Actuators, B, 2018, 267, 476–482 CrossRef CAS.
- Z. Rymansaib, P. Iravani, E. Emslie, M. Medvidović-Kosanović, M. Sak-Bosnar, R. Verdejo and F. Marken, Electroanalysis, 2016, 28, 1517–1523 CrossRef CAS.
- K. Y. Lee, A. Ambrosi and M. Pumera, Electroanalysis, 2017, 29, 2444–2453 CrossRef CAS.
- P. Veluchamy and H. Minoura, J. Mater. Sci. Lett., 1996, 15, 1705–1707 CrossRef CAS.
- G. H. A. Therese and P. V. Kamath, Chem. Mater., 2000, 12, 1195–1204 CrossRef CAS.
- M. E. Snowden, P. H. King, J. A. Covington, J. V. MacPherson and P. R. Unwin, Anal. Chem., 2010, 82, 3124–3131 CrossRef CAS PubMed.
- R. J. Forster, Chem. Soc. Rev., 1994, 23, 289–297 RSC.
- J. Heinze, Angew. Chem., Int. Ed. Engl., 1993, 32, 1268–1288 CrossRef.
- E. A. Hutton, S. B. Hočevar and B. Ogorevc, Anal. Chim. Acta, 2005, 537, 285–292 CrossRef CAS.
- M. A. Nolan and S. P. Kounaves, Anal. Chem., 1999, 71, 3567–3573 CrossRef CAS.
- Y. C. Sun, J. Mierzwa and M. H. Yang, Talanta, 1997, 44, 1379–1387 CrossRef CAS PubMed.
- L. Ramakrishnan, A. Fujishima, D. A. Tryk, M. S. Seehra, J. E. Butler, E. Granite and A. Manivannan, J. Electroanal. Chem., 2005, 577, 287–293 CrossRef.
- L. Landau and B. Levich, Acta Physicochim. URSS, 1942, 17, 141–153 Search PubMed.
- H. Gunasingham and B. Fleet, Anal. Chem., 1983, 55, 1409–1414 CrossRef CAS.
- S. Wang, E. S. Forzani and N. Tao, Anal. Chem., 2007, 79, 4427–4432 CrossRef CAS PubMed.
- J. F. Van Staden and M. C. Matoetoe, Anal. Chim. Acta, 2000, 411, 201–207 CrossRef CAS.
- N. A. Madigan, T. J. Murphy, J. M. Fortune, C. R. S. Hagan and L. A. Coury, Anal. Chem., 1995, 67, 2781–2786 CrossRef CAS.
- Z. Bi, C. S. Chapman, P. Salaün and C. M. G. Van Den Berg, Electroanalysis, 2010, 22, 2897–2907 CrossRef CAS.
- C. S. Chapman and C. M. G. Van Den Berg, Electroanalysis, 2007, 19, 1347–1355 CrossRef CAS.
- W. T. De Vries and E. van Dalen, J. Electroanal. Chem., 1967, 14, 315–327 CrossRef CAS.
- S. J. Cobb and J. V. Macpherson, Anal. Chem., 2019, 91, 7935–7942 CrossRef CAS PubMed.
- J. G. Osteryoung and R. A. Osteryoung, Anal. Chem., 1985, 57, 101A–110A CrossRef CAS.
- R. A. Osteryoung and J. Osteryoung, Philos. Trans. R. Soc., A, 1981, 302, 315–326 CrossRef CAS.
- H. Bagheri, A. Afkhami, H. Khoshsafar, M. Rezaei and A. Shirzadmehr, Sens. Actuators, B, 2013, 186, 451–460 CrossRef CAS.
- H. R. Badiei, J. McEnaney and V. Karanassios, Spectrochim. Acta, Part B, 2012, 78, 42–49 CrossRef CAS.
- Y. Yao, H. Wu and J. Ping, Food Chem., 2019, 274, 8–15 CrossRef CAS PubMed.
- O. A. Farghaly and M. A. Ghandour, Environ. Res., 2005, 97, 229–235 CrossRef CAS PubMed.
- E. A. Hutton, J. T. Van Elteren, B. Ogorevc and M. R. Smyth, Talanta, 2004, 63, 849–855 CrossRef CAS PubMed.
- L. M. Johnson, M. Lynch, C. Johnson and A. B. Karger, Clin. Biochem., 2019, 66, 95–99 CrossRef CAS PubMed.
- A. W. Struempler, Anal. Chem., 1973, 45, 2251–2254 CrossRef CAS.
- J. R. Moody and R. M. Lindstrom, Anal. Chem., 1977, 49, 2264–2267 CrossRef CAS.
- D. P. H. Laxen and R. M. Harrison, Anal. Chem., 1981, 53, 345–350 CrossRef CAS.
- H. B. Ross, Atmos. Environ., 1986, 20, 401–405 CrossRef CAS.
- C. Reimann, A. Grimstvedt, B. Frengstad and T. E. Finne, Sci. Total Environ., 2007, 374, 292–296 CrossRef CAS PubMed.
- K. S. Subramanian, C. L. Chakrabarti, I. S. Maines and J. E. Sueiras, Anal. Chem., 1978, 50, 444–448 CrossRef CAS.
- E. Tipping, Cation Binding by Humic Substances, Cambridge University Press, Cambridge, 1st edn, 2002 Search PubMed.
- T. L. Read, E. Bitziou, M. B. Joseph and J. V. Macpherson, Anal. Chem., 2014, 86, 367–371 CrossRef CAS PubMed.
- T. L. Read, M. B. Joseph and J. V. Macpherson, Chem. Commun., 2016, 52, 1863–1866 RSC.
- Y. Liu, V. Lopez-Avila, J. J. Zhu, D. R. Wiederin and W. F. Beckert, Anal. Chem., 1995, 67, 2020–2025 CrossRef CAS.
- B. Michalke and P. Schramel, Fresenius. J. Anal. Chem., 1997, 357, 594–599 CrossRef CAS.
- X. Jia, Y. Han, X. Liu, T. Duan and H. Chen, Spectrochim. Acta, Part B, 2011, 26, 1380–1386 CAS.
- A. A. Ammann, Anal. Bioanal. Chem., 2002, 372, 448–452 CrossRef CAS PubMed.
- M. Popp, S. Hann and G. Koellensperger, Anal. Chim. Acta, 2010, 668, 114–129 CrossRef CAS PubMed.
- J. J. Berzas Nevado, R. C. Rodríguez Martin-Doimeadios, E. M. Krupp, F. J. Guzmán Bernardo, N. Rodríguez Fariñas, M. Jiménez Moreno, D. Wallace and M. J. Patiño Ropero, J. Chromatogr. A, 2011, 1218, 4545–4551 CrossRef CAS PubMed.
- J. S. Young, J. M. Gurtisen, C. W. Apts and E. A. Crecelius, Mar. Environ. Res., 1979, 2, 265–273 CrossRef CAS.
- P. Pathirathna, T. Siriwardhane, S. P. McElmurry, S. L. Morgan and P. Hashemi, Analyst, 2016, 141, 6432–6437 RSC.
- E. Nieboer and A. Jusys, in Chromium in Natural and Human Environments, ed. J. Nriagu and E. Nieboer, Wiley Interscience, New York, 1988, pp. 21–81 Search PubMed.
- X. C. Le, X. Lu and X.-F. Li, Anal. Chem., 2004, 26A–33A CAS.
- X. Dai, O. Nekrassova, M. E. Hyde and R. G. Compton, Anal. Chem., 2004, 76, 5924–5929 CrossRef CAS PubMed.
- G. Forsberg, J. W. O. Laughlin and R. G. Megargle, Anal. Chem., 1975, 47, 1586–1592 CrossRef CAS.
- K. Pungjunun, S. Chaiyo, I. Jantrahong, S. Nantaphol, W. Siangproh and O. Chailapakul, Microchim. Acta, 2018, 185, 324 CrossRef PubMed.
- L. Pinto and S. G. Lemos, Electroanalysis, 2014, 26, 299–305 CrossRef CAS.
- A. Kawde, A. Ismail, A. R. Al-Betar and O. Muraza, Microporous Mesoporous Mater., 2017, 243, 1–8 CrossRef CAS.
- I. Šinko and J. Doleal, J. Electroanal. Chem., 1970, 25, 299–306 CrossRef.
- Y. K. Chau and K. Lum-Shue-Chan, Water Res., 1974, 8, 383–388 CrossRef CAS.
- J. N. Butler, Carbon Dioxide Equilibria and Their Applications, Routledge, New York, 1st edn, 1991 Search PubMed.
- P. N. Froelic, Limnol. Oceanogr., 1988, 33, 649–668 Search PubMed.
- W. J. Weber and W. Stumm, J. Chem. Eng. Data, 1963, 8, 464–468 CrossRef CAS.
- J. F. Talling, Freshwater Rev., 2010, 3, 133–146 CrossRef.
- C. Song and J. Zhang, in PEM Fuel Cell Electrocatalysts and Catalyst Layers, ed. J. Zhang, Springer London, London, 2008, pp. 89–134 Search PubMed.
- L. Meng, J. Ustarroz, M. E. Newton and J. V. Macpherson, J. Phys. Chem. C, 2017, 121, 6835–6843 CrossRef CAS.
- Y. K. Chau and K. Lum-Shue-Chan, Water Res., 1974, 8, 383–388 CrossRef CAS.
- N. Li, D. Zhang, Q. Zhang, Y. Lu, J. Jiang, G. L. Liu and Q. Liu, Sens. Actuators, B, 2016, 231, 349–356 CrossRef CAS.
- L. A. Hutton, G. D. O'Neil, T. L. Read, Z. J. Ayres, M. E. Newton and J. V. Macpherson, Anal. Chem., 2014, 86, 4566–4572 CrossRef CAS.
- Z. J. Ayres, M. E. Newton and J. V. Macpherson, Analyst, 2016, 141, 3349–3357 RSC.
- G. D. O'Neil, M. E. Newton and J. V. Macpherson, Anal. Chem., 2015, 87, 4933–4940 CrossRef PubMed.
- E. Y. Neiman, L. G. Petrova, V. I. Ignatov and G. M. Dolgopolova, Anal. Chim. Acta, 1980, 113, 277–285 CrossRef CAS.
- M. S. Shuman and G. P. Woodward, Anal. Chem., 1976, 48, 1979–1983 CrossRef CAS.
- G. Zhao and G. Liu, IEEE Sens. J., 2018, 18, 5645–5655 CAS.
- T. M. Florence and G. E. Batley, J. Electroanal. Chem., 1977, 75, 791–798 CrossRef CAS.
- W. Kemula, Z. Galus and Z. Kublik, Nature, 1958, 182, 1228–1229 CrossRef CAS.
- J. Wang, J. Lu, S. B. Hocevar, P. A. M. Farias and B. Ogorevc, Anal. Chem., 2000, 72, 3218–3222 CrossRef CAS PubMed.
- G. E. Batley, Mar. Chem., 1983, 12, 107–117 CrossRef CAS.
- I. Švancara, C. Prior, S. B. Hočevar and J. Wang, Electroanalysis, 2010, 22, 1405–1420 CrossRef.
- H. W. Nurnberg, Fresenius’ Z. Anal. Chem., 1983, 316, 557–565 CrossRef.
- L. Skvortsova, A. Tyo, E. Zakharova and V. Shelkovnikov, in AIP Conference Proceedings, 2016, vol. 1772, pp. 1–9.
- P. Figura and B. McDuffie, Anal. Chem., 1980, 52, 1433–1439 CrossRef CAS.
- M. Wojciechowski, W. Go and J. Osteryoung, Anal. Chem., 1985, 57, 155–158 CrossRef.
- M. Štulíková, Talanta, 1991, 38, 805–807 CrossRef.
- M. Ariel, U. Eisner and S. Gottesfeld, J. Anal. Chem., 1964, 7, 307–314 CAS.
- E. Punrat, S. Chuanuwatanakul, T. Kaneta, S. Motomizu and O. Chailapakul, J. Electroanal. Chem., 2014, 727, 78–83 CrossRef CAS.
- P. L. Brezonik, P. A. Brauner and W. Stumm, Water Res., 1976, 10, 605–612 CrossRef CAS.
- W. W. Kubiak and J. Wang, Talanta, 1989, 36, 821–824 CrossRef CAS PubMed.
- Y. Bonfil and E. Kirowa-Eisner, Anal. Chim. Acta, 2002, 457, 285–296 CrossRef CAS.
- J. Wang and J. M. Zadeii, J. Electroanal. Chem., 1988, 246, 297–305 CrossRef CAS.
- R. B. McCleskey, D. Kirk Nordstrom and J. N. Ryan, Appl. Geochem., 2011, 26, S227–S229 CrossRef CAS.
- J. R. Gray, Environ. Instrum. Anal. Handb., 2004, 491–510 Search PubMed.
- a J. deBethune, T. S. Licht and N. Swendeman, J. Electrochem. Soc., 1959, 106, 616–625 CrossRef CAS.
- S. B. Hočevar, B. Ogorevc, J. Wang and B. Pihlar, Electroanalysis, 2002, 14, 1707–1712 CrossRef.
- W. Lund and R. Eriksen, Anal. Chim. Acta, 1979, 107, 37–46 CrossRef CAS.
- F. Marken, S. L. Matthews, R. G. Compton and B. A. Coles, Electroanalysis, 2000, 12, 267–273 CrossRef CAS.
- G. Aragay, J. Pons and A. Merkoçi, J. Mater. Chem., 2011, 21, 4326–4331 RSC.
- M. Jasinski, A. Kirbs, M. Schmehl and P. Grundler, Electrochem. Commun., 1999, 1, 26–28 CrossRef CAS.
- B. A. Coles, Y.-C. Tsai, K. Holt, F. Marken, R. G. Compton and J. S. Foord, Electroanalysis, 2002, 13, 831–835 Search PubMed.
- R. S. Sadana, Anal. Chem., 1983, 55, 304–307 CrossRef CAS.
- G. E. Batley and D. Gardner, Estuarine Coastal Mar. Sci., 1978, 7, 59–70 CrossRef CAS.
- C. K. Jain and D. Ram, Hydrol. Sci. J., 1997, 42, 713–723 CrossRef CAS.
- N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, J. Chem. Educ., 2018, 95, 197–206 CrossRef CAS.
- L. A. Hutton, J. G. Iacobini, E. Bitziou, R. B. Channon, M. E. Newton and J. V. Macpherson, Anal. Chem., 2013, 85, 7230–7240 CrossRef CAS PubMed.
- G. Kefala, A. Economou and A. Voulgaropoulos, Analyst, 2004, 129, 1082–1090 RSC.
- J. Wang, R. P. Deo, S. Thongngamdee and B. Ogorevc, Electroanalysis, 2001, 13, 1153–1156 CrossRef CAS.
- M. Lizotte, C. Hoffman, D. Lechleiter and J. McDonald, US Pat., US6779383B2, 2004 Search PubMed.
Footnote |
| † Both authors contributed equally to this article. |
| This journal is © The Royal Society of Chemistry 2019 |