
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solvent-independent determination of heteroatom protonation states from NMR spectra by differential deuterium isotope shifts†
Sebastian
Tassoti‡
 a,
Martin
Walenta‡
a,
Alexander
Pöcheim
a,
Kathrin
Buchberger
a,
Olaf
Kunert
b and
Klaus
Zangger
*a
a,
Martin
Walenta‡
a,
Alexander
Pöcheim
a,
Kathrin
Buchberger
a,
Olaf
Kunert
b and
Klaus
Zangger
*a
aInstitute of Chemistry/Organic and Bioorganic Chemistry, University of Graz, Austria. E-mail: klaus.zangger@uni-graz.at
bInstitute of Pharmaceutical Sciences, University of Graz, Austria
First published on 11th November 2019
Abstract
The NMR-spectroscopy based structure elucidation of organic molecules containing heteroatoms is often obstructed by the difficulties in determining the heteroatom protonation states. Here we describe a simple but broadly applicable approach for the determination of the protonation states of heteroatoms. Differential deuterium isotope shifts observed upon the addition of small amounts of H2O or D2O to any solvent can be used to determine the protonation states of heteroatoms.
Introduction
NMR spectroscopy is one of the techniques most often used for the structure elucidation of small organic molecules as well as biomolecules. However, the determination of the protonation states of heteroatoms such as oxygen, nitrogen, sulphur and phosphorus is frequently hindered by two factors. First, protons of protonated heteroatoms are often not visible due to exchange broadening1–6 and second, even if there is no broadening, multi-bond correlations to a carbon nucleus are not always found easily in multi-bond correlation experiments.7 For example, it is often difficult to distinguish alcohol functions from ether moieties. To overcome this limitation, partial NMR solutions have been presented.Since the early days of solution-state NMR spectroscopy, the substitution of hydrogen atoms by deuterium has been thoroughly studied.7–14 Upon replacement of hydrogen by deuterium in a molecule, the dynamic state of this molecule is changed. Therefore, the electronic structure is modified, which in turn leads to an alteration of the magnetic shielding in the vicinity of the replaced proton.15,16 The magnitude of the observed isotope shift for a given carbon nucleus depends on its distance from the site of isotopic substitution as well as the number of deuterium atoms substituting hydrogen.17 This was used in early reports to identify a broad spectrum of functional groups in compounds like peptides,17,18 carbohydrates,19,20 enaminones,21 phenols22 and other alcohols.23
Experimentally, the deuteration of the compounds was achieved using different approaches. For example, the partial or full deuteration of compounds of interest by dissolution in deuterium oxide, followed by incubation for three days, a simple but time-consuming process.17,24 In a more elaborate approach, the use of a coaxial dual-cell setup was proposed, where the inner tube contains the analyte dissolved in water, whereas in the outer tube it is dissolved in deuterium oxide.19,20,23 When recording NMR spectra within this setup, the differential chemical shifts in water vs. the deuterium oxide are obtained simultaneously. However, this method is limited to water-soluble compounds. Another approach for the simultaneous determination of the deuterated and the hydrogenated form of the compound is the dissolution of an equimolar mixture of the deuterated and the normal compound.22 While this approach overcomes the solubility issues stated, the sample preparation procedure is time- and cost-consuming.
Further observations of deuterium-induced effects include their influence on hydrogen-bonded complexes,25 ions26 and tautomeric equilibria.27 For additional information on the application of deuterium isotope shifts, there are dedicated reviews available.28–30
Here we present a fast, simple and generally applicable approach for the determination of the protonation states of heteroatoms in organic compounds. Thereby, the isotopic shift introduced by deuteration of heteroatoms, which is in turn detected by 13C or 31P NMR is obtained in any desired solvent in the presence of 10% water or 10% deuterium oxide. A wide range of solvents can be employed with this method, without the prerequisite of miscibility with water. Furthermore, there is no need for deuteration of the compound during sample preparation. Additionally, deuterated solvents can be used without prior drying.
Results and discussion
For the discrimination of the protonation states of heteroatoms, two 13C-NMR spectra were recorded after the addition of water or deuterium oxide and the chemical shifts obtained for all nuclei were compared after referencing the spectra with TMS. From these chemical shift values, a differential isotope shift (DIS) value was calculated, describing the difference of the chemical shift in the presence of deuterium oxide versus protonated water.| DIS = δ(H2O) − δ(D2O) |
The differential isotope shift value of nuclei depends on their neighbouring atoms, specifically if they are close to exchangeable protons, but is temperature-independent. Employing the presented method, these protons are readily replaced by deuterium originating from deuterated water. In the following, the DIS-values in deuterated methanol will be presented as an example. A comparison of different solvents can be found in Table 1 and data for all investigated solvents are presented in the ESI.† As a first step, we characterized the DIS-values of carbon nuclei near oxygen atoms, which were either alcohols, ethers, esters or ketones. For alcohols, where protons bound to heteroatoms are swiftly exchanged by deuterium in the presence of deuterium oxide, the DIS-value of carbon nuclei close-by the deuterated heteroatom are positive, because the atoms experience a significant upfield shift. The α-carbon next to the oxygen atom in question shows a high DIS-value, typically around 0.1 ppm, if the oxygen is protonated (see compounds 1 through 3), and hence deuterated in the presence of deuterium oxide, whereas the β-carbon nucleus experiences a shift dictated by the chemical environment (compare 1–3). In contrast, oxygen atoms which cannot be protonated, like ketones (4), esters (5) or ethers (6) show small DIS-values around zero close to the heteroatom, as the precision of the method is around 0.01 ppm (Fig. 1).
 | ||
| Fig. 1 Various oxygen containing compounds and their corresponding deuterium isotope shift values of each carbon atom (recorded in deuterated methanol, DIS value is given as ppm). Here, a higher DIS value (>0.05 ppm) points to a carbon atom close or next to a hydroxyl group. | ||
| MeOD-d4 | Pyridine-d5 | CDCl3 | |
|---|---|---|---|
| Alcohol | 0.10 ± 0.02 | 0.12 ± 0.01 | 0.15 ± 0.02 |
| Ether | 0.00 ± 0.01 | 0.00 ± 0.01 | 0.00 ± 0.02 |
| Primary amine | 0.14 ± 0.04 | 0.17 ± 0.03 | 0.17 ± 0.04 |
| Secondary amine | 0.07 ± 0.02 | 0.10 ± 0.01 | 0.10 ± 0.04 |
| Tertiary amine | 0.00 ± 0.01 | 0.00 ± 0.01 | 0.00 ± 0.01 |
| Thiol | 0.16 ± 0.02 | 0.17 ± 0.03 | 0.18 ± 0.03 |
| Thioether | 0.01 ± 0.02 | 0.00 ± 0.01 | 0.00 ± 0.02 |
These observations are also true for compounds where more than one heteroatom is present. It reliably distinguishes alcohols from ketones, esters and ethers even in multi-functionalized compounds (see 7, 8 and 10 in Fig. 2). There is also a high tolerance for structural elements like aromatic rings and heterocycles as demonstrated in Fig. 2. For example, when two or more ether groups and one alcohol function are present, the carbon next to the alcohol possesses the highest DIS value, in an open chain as well as in a diacetal-protected sugar molecule (10). However, it is not possible to distinguish ethers, esters or ketones from each other, because their DIS-values are not characteristic for the respective groups (9).
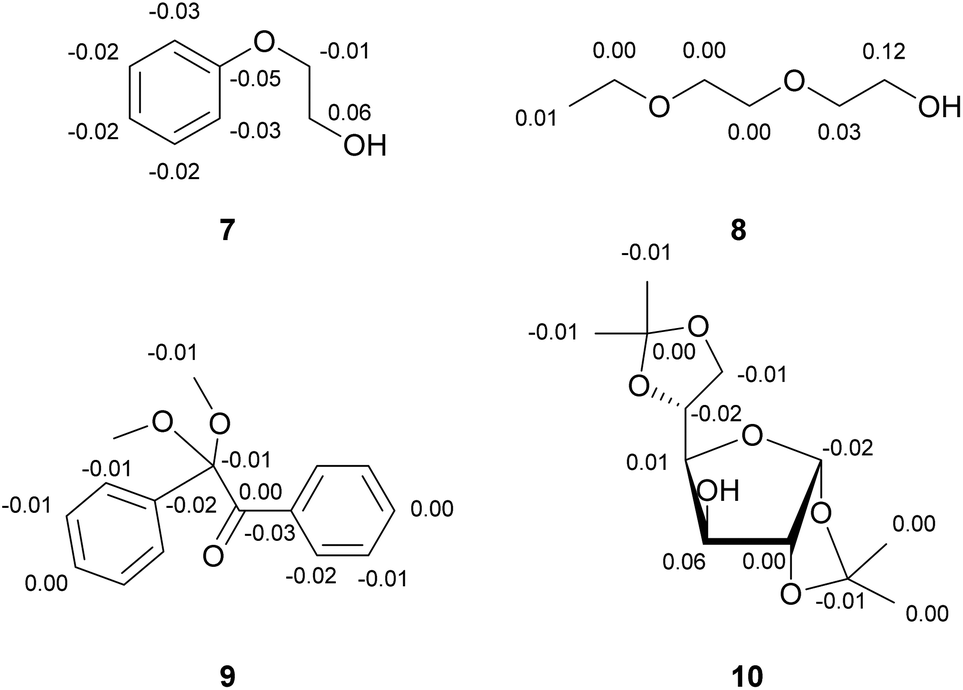 | ||
| Fig. 2 Compounds containing multiple oxygen atoms still produce reliable DIS data (recorded in deuterated methanol, DIS-value in ppm). A positive shift can be attributed to proximity to an alcohol moiety. | ||
Similar to oxygen, the protonation state of other heteroatoms such as nitrogen or sulphur can be determined employing this method. The same rules apply, where a primary amine can be distinguished from a tertiary amide or amine (11–13) (see Fig. 3). Likewise, thiols are different from thioethers and sulphur-containing heterocycles (14, 15). Again, there is a high tolerance for several structural elements (Fig. 3).
 | ||
| Fig. 3 Nitrogen- and sulphur-containing compounds and their corresponding DIS-values (as determined in methanol, DIS-value in ppm). | ||
When multiple heteroatoms are present in a compound (see Fig. 4), the employed method can generally distinguish between protonated and non-protonated moieties. For example, a thioether can be discriminated from a primary amine (16). In case of two different protonated heteroatoms, such as sulphur and oxygen, a tendency towards higher DIS-values next to thiols in contrast to slightly lower DIS-values nearby alcohol-groups can be observed (17, 18). In the case of presence of three different protonated heteroatoms in the same molecule, we observed the highest deuterium isotope shift for a thiol, followed by a secondary amide group and a carboxylic acid (19). Generally, we have observed this trend in all solvents tested (see Table 1). In methanol, pyridine and chloroform the DIS-values depend on the heteroatom, for example thiols show larger DIS-values than amines or alcohols. However, the numeric value can only be used as a first indication of which heteroatom is present in a compound and not as an unambiguous assignment.
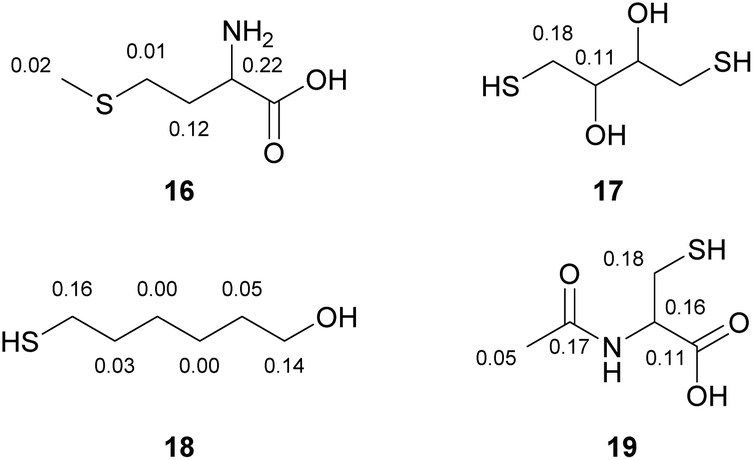 | ||
| Fig. 4 Deuterium isotope shift values (recorded in deuterated methanol, DIS-values in ppm) of compounds containing several heteroatoms in different protonation states. | ||
Elaborating on this concept, deuterium isotope shifts are not only of great importance in carbon NMR spectroscopy. For example, the discrimination of mono-, bis- and tris-alkylated phosphate esters is of high interest. However, the grade of substitution cannot easily be determined by NMR, due to the fact that the chemical shift of phosphorus in alkyl phosphates is close to 0 ppm for mono-, di- and tri-esters. Using our sample preparation method in conjunction with 31P NMR spectroscopy, we were able to distinguish tris(2-ethylhexyl) phosphate (22) from its mono- (20) and bi-substituted (21) forms. These two compounds show a positive DIS-value, which is in contrast to the tri-substituted ester, where no deuterium isotope shift can be observed (see Fig. 5).
 | ||
| Fig. 5 Differentiation of mono- and di- from tri-phosphate-esters. DIS-values were observed by phosphorus NMR spectroscopy and measured in deuterated ACN. No deuterium isotope shift is observed for the tri-substituted compound. | ||
To demonstrate the power of the presented approach on more complex systems containing multiple heteroatoms it has been employed on the macrolide antibiotic erythromycin. In erythromycin (23), the chemical shifts of carbons neighbouring the five hydroxyl and the five ether groups lie within a very narrow spectral range.31 Long-range heteronuclear correlations are not guaranteed to be seen,32 and therefore the distinction of ether groups from alcohols is not trivial. Employing our method, we were able to swiftly identify the carbon nuclei neighbouring hydroxy-moieties, which possess a deuterium isotope shift greater than 0.10 ppm. In contrast, the DIS-value near ether groups is between 0.00 ppm and 0.03 ppm (see Fig. 6).
 | ||
| Fig. 6 DIS-values found in erythromycin (23) and rutin (24). For clarity, only deuterium isotope shifts near heteroatoms are given. | ||
Yet another example of the capabilities of the presented method is the assignment of rutin (24), a flavone glycoside with a saccharide chain consisting of two moieties. This molecule comprises several ether and hydroxyl groups, of which nine neighbour a carbon atom with a signal in a very narrow spectral range. Without elaborate two-dimensional experiments no unambiguous assignment can be made. Using DIS-values, as shown in Fig. 6 we can quickly differentiate between the alcohol and ether groups and facilitate the assignment with a fast and simple experiment.
The results for rutin also demonstrate that the same DIS-values can be obtained with a concentration of water of only 2%. This low concentration facilitates assignments of the NMR resonances of the water containing sample by comparison with NMR shift values of a water free sample of the same compound.
The presented approach can be used for assessing the plausibility of assignments in complex saccharide chains as carbon atoms at points of fusion should not be affected by change of water to deuterated water.
However, the major advantage of DIS-values is the ability to determine free OH groups in novel natural compounds, which often had to be chemically modified (acetylated, methylated) in the past in order to determine free OH groups and differentiate isomeric compounds. With the presented approach it is possible to get the same information without consumption of precious biological material.
Experimental
All chemicals were purchased from Sigma-Aldrich at >98% purity. Approximately 20 mg of the sample were dissolved in 500 μL of deuterated solvent (pyridine, DMSO, acetone, acetonitrile, chloroform or methanol). For protonation/deuteration of heteroatoms, 50 μL of water or deuterated water were added. TMS was added for chemical shift referencing. In the case of methanol, methanol-d3 was used for samples with water and methanol-d4 was used for the samples with deuterated water. For the phosphate esters, ACN-d3 was used as solvent and triphenylphosphate was added for chemical shift referencing. All samples were measured in duplicates. In case of the flavonol glycoside rutin, 5 mg were dissolved in 600 μL pyridine-d5 and 10% or 2% of water or deuterated water were added. The experiments were recorded on a Bruker Avance III 700 MHz spectrometer at 298 K.All other NMR spectra were recorded on a Bruker AVANCE III 300 MHz spectrometer or on a Bruker Avance III 500 MHz spectrometer using a 5 mm TXI probe with z-axis gradients at 300 K. Typically, 512 scans and 32k data points were acquired for proton decoupled carbon-spectra. Spectra were processed using MestreNova software.
Conclusions
In conclusion, we have developed a simple yet versatile method for the determination of the protonation state of oxygen, nitrogen, sulphur and phosphorus in organic compounds. It is applicable in a range of solvents and is robust to many functional groups. It comprises the acquisition of NMR spectra in which the chemical shift of the α-atom of the heteroatom in question is observed both in the presence of water and in the presence of deuterium oxide. The presented method is capable of the discrimination of the protonation state of the heteroatom by the determination of the deuterium isotope shift and is a valuable tool in the structure determination of novel natural compounds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support to K. Z. by the Austrian Science Foundation (FWF) under project number P30230 and through the Doktoratskolleg Molecular Enzymology (W901) is gratefully acknowledged. We also thank the interuniversity program in natural sciences, NAWI Graz, for financial support.Notes and references
- H. S. Gutowsky and C. H. Holm, J. Chem. Phys., 1956, 25, 1228–1234 CrossRef CAS.
- J. T. Arnold, Phys. Rev., 1956, 102, 136–150 CrossRef CAS.
- H. S. Gutowsky and A. Saika, J. Chem. Phys., 1953, 21, 1688–1694 CrossRef CAS.
- E. L. Hahn and D. E. Maxwell, Phys. Rev., 1952, 88, 1070–1084 CrossRef CAS.
- H. S. Gutowsky, D. W. McCall and C. P. Slichter, J. Chem. Phys., 1953, 21, 279–292 CrossRef CAS.
- J. Kaplan, J. Chem. Phys., 1958, 28, 278–282 CrossRef CAS.
- A. Bax and M. F. Summers, J. Am. Chem. Soc., 1986, 108, 2093–2094 CrossRef CAS.
- J. Stothers, C. Tan, A. Nickon, F. Huang, R. Sridhar and R. Weglein, J. Am. Chem. Soc., 1972, 94, 8581–8582 CrossRef CAS.
- A. P. Tulloch, Can. J. Chem., 1977, 55, 1135–1142 CrossRef CAS.
- P. A. J. Gorin, Can. J. Chem., 1974, 52, 458–461 CrossRef CAS.
- B. Coxon, Carbohydr. Res., 2005, 340, 1714–1721 CrossRef CAS PubMed.
- T. Dziembowska, P. E. Hansen and Z. Rozwadowski, Prog. Nucl. Magn. Reson. Spectrosc., 2004, 45, 1–29 CrossRef CAS.
- M. Takeda, Y. Miyanoiri, T. Terauchi, C.-J. Yang and M. Kainosho, J. Magn. Reson., 2014, 241, 148–154 CrossRef CAS PubMed.
- S. Hanashima, K. Kato and Y. Yamaguchi, Chem. Commun., 2011, 47, 10800 RSC.
- H. Batiz-Hernandez and R. A. Bernheim, Prog. Nucl. Magn. Reson. Spectrosc., 1967, 3, 63–85 CrossRef CAS.
- L. J. Altman, P. Laungani, G. Gunnarsson, H. Wennerstrom and S. Forsen, J. Am. Chem. Soc., 1978, 100, 8264–8266 CrossRef CAS.
- H. K. Ladner, J. J. Led and D. M. Grant, J. Magn. Reson., 1975, 20, 530–534 CAS.
- J. Feeney, P. Partington and G. C. Roberts, J. Magn. Reson., 1974, 13, 268–274 CAS.
- P. E. Pfeffer, K. M. Valentine and F. W. Parrish, J. Am. Chem. Soc., 1979, 101, 1265–1274 CrossRef CAS.
- P. E. Pfeffer, F. W. Parrish and J. Unruh, Carbohydr. Res., 1980, 84, 13–23 CrossRef CAS.
- D. K. Zheglova, D. G. Genov, S. Bolvig and P. E. Hansen, Acta Chem. Scand., 1997, 51, 1016–1023 CrossRef CAS.
- R. A. Newmark and J. R. Hill, Org. Magn. Reson., 1980, 13, 40–44 CrossRef CAS.
- B. Capon, D. S. Rycroft, T. W. Watson and C. Zucco, J. Am. Chem. Soc., 1981, 103, 1761–1765 CrossRef CAS.
- J. C. Christofides and D. B. Davies, J. Am. Chem. Soc., 1983, 105, 5099–5105 CrossRef CAS.
- S. N. Smirnov, N. S. Golubev, G. S. Denisov, H. Benedict, P. Schah-Mohammedi and H. H. Limbach, J. Am. Chem. Soc., 1996, 118, 4094–4101 CrossRef CAS.
- P. Schah-Mohammedi, I. G. Shenderovich, C. Detering, H.-H. Limbach, P. M. Tolstoy, S. N. Smirnov, G. S. Denisov and N. S. Golubev, J. Am. Chem. Soc., 2000, 122, 12878–12879 CrossRef CAS.
- Y. Zhu, J. Zajicek and A. S. Serianni, J. Org. Chem., 2001, 66, 6244–6251 CrossRef CAS PubMed.
- P. E. Hansen, Magn. Reson. Chem., 2000, 38, 1–10 CrossRef CAS.
- S. Bolvig and P. E. Hansen, Curr. Org. Chem., 2000, 4, 19–54 CrossRef CAS.
- T. Dziembowska and Z. Rozwadowski, Curr. Org. Chem., 2001, 5, 289–313 CrossRef CAS.
- J. Barber, J. I. Gyi, L. Lian, G. A. Morris, D. A. Pye and J. K. Sutherland, J. Chem. Soc., Perkin Trans. 2, 1991, 1489–1494 RSC.
- P. Tyson, A. Hassanzadeh, M. N. Mordi, D. G. Allison, V. Marquez and J. Barber, MedChemComm, 2011, 2, 331–336 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9an01364d |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2019 |