
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Analysis of long dsRNA produced in vitro and in vivo using atomic force microscopy in conjunction with ion-pair reverse-phase HPLC†
Alison O.
Nwokeoji‡
a,
Sandip
Kumar‡
b,
Peter M.
Kilby
c,
David E.
Portwood
c,
Jamie K.
Hobbs
*b and
Mark J.
Dickman
 *a
*a
aDepartment of Chemical and Biological Engineering, Mappin Street, University of Sheffield, S1 3JD, UK. E-mail: m.dickman@sheffield.ac.uk
bDepartment of Physics and Astronomy, Hounsfield Road, University of Sheffield, S3 7RH, UK. E-mail: jamie.hobbs@sheffield.ac.uk
cSyngenta, Jealott's Hill International Research Centre, Bracknell, Berkshire RG42 6EY, UK
First published on 22nd July 2019
Abstract
Long double-stranded (ds) RNA is emerging as a novel alternative to chemical and genetically-modified insect and fungal management strategies. The ability to produce large quantities of dsRNA in either bacterial systems, by in vitro transcription, in cell-free systems or in planta for RNA interference applications has generated significant demand for the development and application of analytical tools for analysis of dsRNA. We have utilised atomic force microscopy (AFM) in conjunction with ion-pair reverse-phase high performance liquid chromatography (IP-RP-HPLC) to provide novel insight into dsRNA for RNAi applications. The AFM analysis enabled direct structural characterisation of the A-form duplex dsRNA and accurate determination of the dsRNA duplex length. Moreover, further analysis under non-denaturing conditions revealed the presence of heterogeneous dsRNA species. IP-RP-HPLC fractionation and AFM analysis revealed that these alternative RNA species do not arise from different lengths of individual dsRNA molecules in the product, but represent misannealed RNA species that present as larger assemblies or multimeric forms of the RNA. These results for the first time provide direct structural insight into dsRNA produced both in vivo in bacterial systems and in vitro, highlighting the structural heterogeneity of RNA produced. These results are the first example of detailed characterisation of the different forms of dsRNA from two production systems and establish atomic force microscopy as an important tool for the characterisation of long dsRNA.
1. Introduction
The application of dsRNA for the targeted, sequence specific inhibition of specific genes via RNA interference (RNAi) is emerging as an important tool for the development of novel RNAi-based sustainable insect and fungal management strategies.1–3 There are a wide range of future potential applications of dsRNA based biocontrols for agricultural insect pests as well as for prevention of diseases in beneficial insects. The production of large quantities of dsRNA using a variety of methods including in vitro transcription, bacterial systems, cell-free systems or in planta for RNA interference requires a variety of robust analytical techniques to fully characterise and accurately quantify dsRNA prior to RNAi applications.Most RNAi research in insects has been performed using dsRNA constructs of between 100–800 bp![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 4–6 and a minimum length of approximately 60 bp for effective triggering of RNAi has been demonstrated in several insects.7–9 The use of longer dsRNA molecules generates many siRNAs via dicer cleavage, which contributes to the RNAi response and prevents the resistance due to polymorphic variation in nucleotide sequences.
4–6 and a minimum length of approximately 60 bp for effective triggering of RNAi has been demonstrated in several insects.7–9 The use of longer dsRNA molecules generates many siRNAs via dicer cleavage, which contributes to the RNAi response and prevents the resistance due to polymorphic variation in nucleotide sequences.
Large scale synthesis of dsRNA for RNAi applications, including the production of RNA-based insecticides and fungicides has created a demand for biochemical and biophysical techniques to analyse the long dsRNA produced. Such analytical approaches are aimed at the characterisation (including analysis of the structural homogeneity of the dsRNA) prior to downstream applications.
AFM has been widely and more routinely used for the analysis of dsDNA, including the imaging of short and long dsDNA and a number of dsDNA–protein complexes.10–13 However, there are only limited, more recent applications of AFM for the analysis of RNA and RNA–protein complexes.14 The structural heterogeneity associated with functional biological RNA molecules, in conjunction with the difficulties associated with synthesis and purification of intact RNA in the absence of deleterious ribonucleases that degrade RNA have limited the opportunities for the biophysical and structural analysis. Previously AFM was applied to image viral dsRNA to calculate its length.15 More recently, structural properties of dsRNA were compared to those of dsDNA using AFM in air16 and high-resolution AFM in liquid was able to image the helical pitch of an in vitro synthesized dsRNA.17
AFM approaches have also been applied to study and provide further structural insight into more complex ssRNA assemblies and multimers in a number of important biological RNAs including the analysis of ribozymes,18 the HIV-1Rev response element (RRE) RNA (including analysis of the complex with Rev).19 In addition, AFM has been widely used for imaging a wide variety of different RNA nanostructures in the emerging RNA nanotechnology field.20
In this study we have used ion pair reverse phase chromatography (IP RP HPLC) under both non-denaturing and denaturing conditions in conjunction with high resolution AFM imaging to characterise long dsRNA and their associated dsRNA multimers or higher order assemblies generated both in vitro and in vivo in E. coli for use as RNA-based insecticides.
2. Experimental methods
2.1. Materials and reagents
Genes were synthesised by GeneArt Gene Synthesis (ThermoFisher Scientific). Ampicillin sodium salt, tetracycline hydrochloride, isopropyl β-D-1-thiogalactopyranoside (IPTG) ≥99%, sodium dodecyl sulphate (SDS), sodium chloride (NaCl), dimethyl sulfoxide (DMSO), poly-L-ornithine, Luria Broth (LB) were all obtained from Sigma Aldrich. HPLC grade water, acetonitrile and isopropanol were obtained from Fisher Scientific.2.2. Expression and purification of dsRNA in vivo using E. coli HT115 (DE3)
The E. coli strain, HT115 (DE3)21 was obtained from Cold Spring Harbor Laboratory, NY, USA. Plasmids pDome11 and pCOIV contained in-house designed sequences flanked on both sides with T7 promoters and T7 terminators. The genes were synthesised by GeneArt (ThermoFisher) and cloned into pMX cloning vectors. Theoretical sizes of the dsRNA assuming run through transcription are 521 and 698 base pairs. The pDome11 and pCOIV transformed cells were grown in Luria Broth (LB) and induced with IPTG to express dsRNAs as previously described.22 RNA purification was performed using the RNASwift method as previously described.23 Full details are provided in ESI.†RNA quantification was performed using a Nanodrop 2000 UV visible spectrophotometer (Thermo Fisher Scientific) using an extinction coefficient of 0.021 (μg mL−1)−1 cm−1 which corresponds to 1 A260 = 46.52 μg ml−1.24
2.3. Ion-pair reverse-phase high performance liquid chromatography (IP RP HPLC)
Samples were analysed by IP-RP-HPLC on a passivated Agilent 1100 series HPLC using a Proswift RP-1S Monolith column (50 mm × 4.6 mm I.D. ThermoFisher). Chromatograms were generated using UV detection at a wavelength of 260 nm. The chromatographic analysis was performed using the following conditions: buffer A 0.1 M triethylammonium acetate (TEAA) pH 7.0 (Fluka, UK); buffer B 0.1 M TEAA, pH 7.0 containing 25% acetonitrile (ThermoFisher). RNA was analysed using the following gradient. The gradient started at 22% buffer B to 27% in 2 minutes, followed by a linear extension to 62% buffer B over 15 minutes, then to 73% buffer B over 2.5 minutes at a flow rate of 1.0 ml min−1 at either 50 °C or 80 °C.2.4. In vitro transcription (IVT) of dsRNA and ssRNA
For dsRNA synthesis via in vitro transcription, two DNA templates for the sense and anti-sense transcripts were used resulting in a 504 nt transcribed sequence. The complementary ssRNA transcripts were then generated using in vitro transcription in conjunction with HiScribe™ T7 High Yield RNA Synthesis Kit (New England Biolabs): 10 mM NTPs, 1× reaction buffer, 1 μg DNA template and 2 μl HiScribe™ T7 polymerase in 20 μl RNase-free water. To synthesize the dsRNA, the complementary ssRNA transcripts were mixed in equal proportions and annealed at room temperature. To generate dsDNA, pBluesciptII KS(+) plasmid was used as a template to generate a 228 bp dsRNA using PCR. Nucleic acid compositions of the dsRNA and dsDNA are shown in ESI Table S1.†2.5. Atomic force microscopy
The double stranded nucleic acid polymers, dsRNA and dsDNA, were immobilised on poly-L-ornithine coated mica and imaged in water or air using Tapping Mode™ AFM imaging. 50 μl of 10 μg ml−1 of poly-L-ornithine (molecular weight 30000–70000, Sigma-Aldrich, P3655) was deposited on freshly cleaved 12 mm ‘Ruby muscovite mica’ discs (Agar Scientific, AGG250). After approximately 5 minutes, 40 μl of the solution was removed from the drop. The mica surface was then rinsed 4 times with 100 μl water (HPLC grade) and dried in nitrogen flow giving a uniform coverage of poly-L-ornithine on the mica. 10 μl of 0.1 to 1 ng μl−1 of DNA or RNA diluted in 10 mM Tris-HCl pH 8.0 buffer was deposited on the freshly prepared poly-L-ornithine coated mica. The drop was left for 5 minutes and then rinsed 4 times with 100 μl water and imaged in water. After imaging, the same sample was dried in a nitrogen flow and imaged in air. All the imaging was performed in Tapping Mode™ using a Dimension FastScan Bio AFM (Bruker). The tip was engaged at a free amplitude of about 50 mV (the minimum value allowed by the software to engage). While engaged, the Z-range was reduced to improve signal to noise,25 the free amplitude was lowered to reduce the imaging force, and the feedback gains were optimised to ensure tracking of the nucleic acid polymers without moving them. For imaging in water, FastScanD-SS (Bruker) cantilevers with a silicon tip (nominal values: tip radius 1 nm, cantilever length 15 μm, stiffness 0.25 N m−1, resonant frequency 100 kHz in liquid) were tuned to about 80 kHz, oscillated at a free amplitude of <50 mV (<1 nm) and scanned at a tip velocity of 1–4 μm s−1. For the high resolution images in Fig. 1(A, B) and 2(B), USC-F1.2-k0.15 cantilevers (NanoWorld) with a High Density Carbon/Diamond Like Carbon tip (nominal values: tip radius 10 nm, cantilever length 7 μm, stiffness 0.15 N m−1, resonant frequency 1200 kHz in air) were tuned to 600–650 kHz, oscillated at a free amplitude of <30 mV and scanned at a rate of 0.4–1.0 μm s−1, to visualize the dsRNA and dsDNA grooves. For imaging in air, TESPA-V2 (Bruker) cantilevers with a silicon tip (nominal values: tip radius 7 nm, cantilever length 123 μm, stiffness 37 N m−1, resonant frequency 320 kHz in air) were auto-tuned to just below the resonant frequency and the sample was imaged at a free amplitude of 30 mV (∼3 nm) and a scan rate of 2 μm s−1.
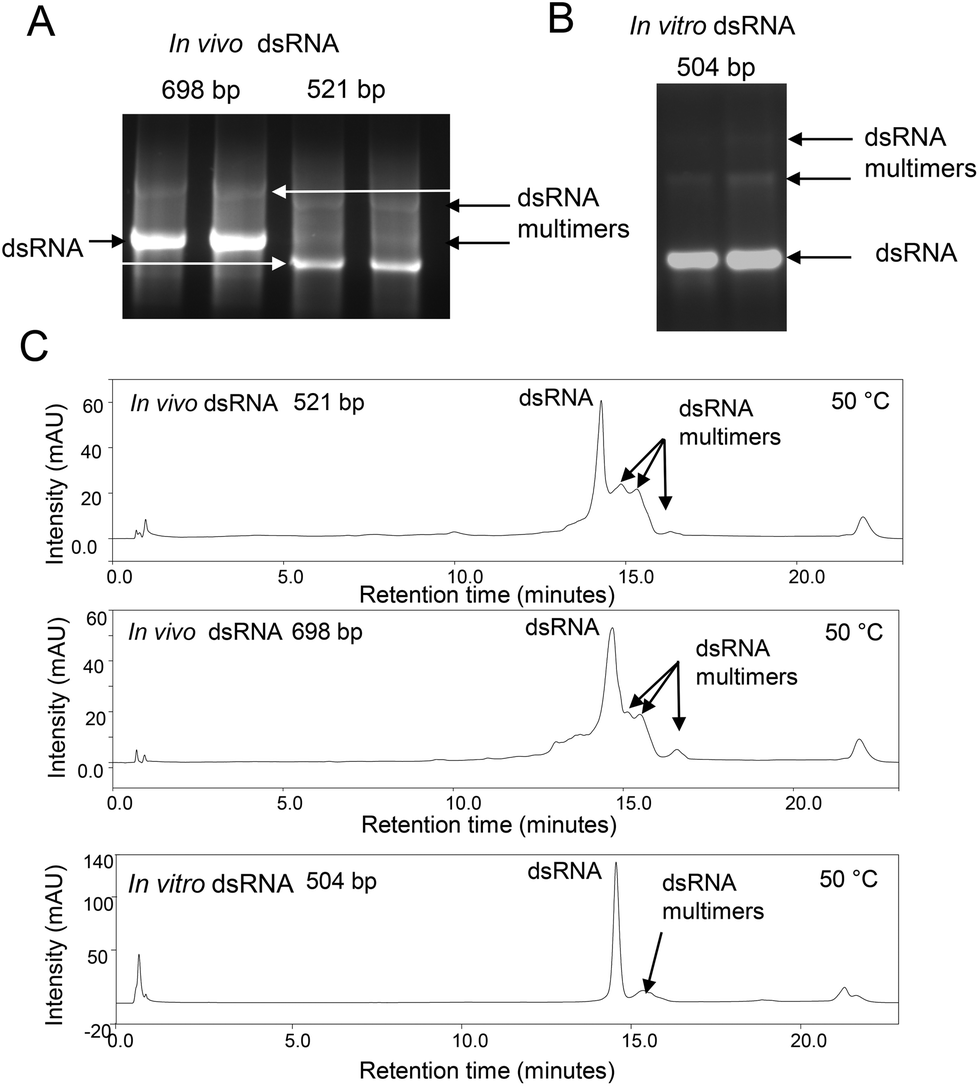 | ||
| Fig. 1 Analysis of dsRNA monomers, multimers and higher order assemblies under non-denaturing conditions. Non-denaturing gel electrophoretograms (A) in vivo synthesised dsRNA (521 bp and 698 bp) (B) in vitro synthesised dsRNA (504 bp). Each dsRNA sample was run in duplicate. The proposed dsRNA multimers or higher order assemblies with reduced electrophoretic mobility are highlighted above the corresponding dsRNA main band. (C) Non denaturing IP-RP-HPLC (50 °C) of in vivo synthesised dsRNA (521 bp and 698 bp) and in vitro synthesised dsRNA (504 bp). The proposed dsRNA multimers with increased retention times are highlighted. | ||
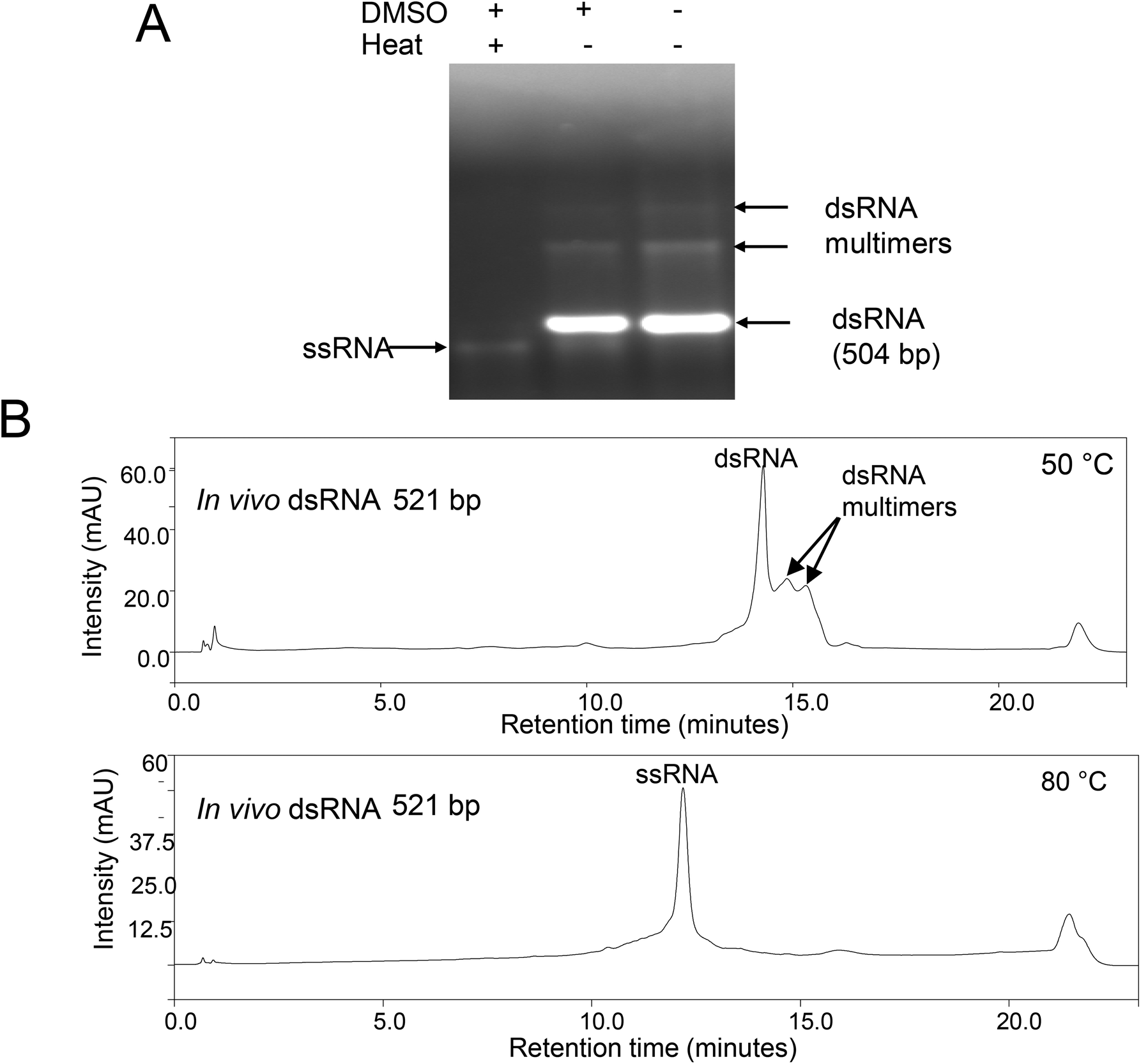 | ||
| Fig. 2 Analysis of dsRNA multimers or higher order assemblies under denaturing conditions. (A) In vitro synthesised 504 bp dsRNA was incubated in the presence and absence of 50% DMSO with or without a short thermal denaturation step prior to gel electrophoresis. The corresponding ssRNA, dsRNA and dsRNA multimers or higher order assemblies are shown. (B) Non-denaturing (50 °C) and denaturing (80 °C) IP-RP-HPLC of in vivo synthesised 521 bp dsRNA. The proposed dsRNA multimers or higher order assemblies with increased retention times are highlighted. The corresponding ssRNAs are shown. | ||
3. Results and discussion
3.1. Analysis of long dsRNA produced in vitro and in vivo reveals heterogeneous products
Large quantities of dsRNA can be readily synthesised in either bacterial systems, by in vitro transcription, in cell-free systems or in planta in order to specifically block the expression of essential genes, resulting in insect mortality. It is important to characterise the dsRNA generated prior to use as RNA-based insecticides. In this study we focused on the production of dsRNA using both in vitro transcription and transcription in E. coli (generated in vivo) by growing E. coli HT115 cells transformed with plasmids to express dsRNA. Following E. coli growth, dsRNA was extracted using conditions to ensure all ssRNA is degraded, prior to purification of the dsRNA using solid phase extraction (SPE).23 For in vitro transcription of dsRNA the corresponding ssRNAs were in vitro transcribed prior to purification using SPE and formation of dsRNA by incubating equal concentrations of the ssRNA. The analyses of dsRNA using native agarose gel electrophoresis following extraction and purification of dsRNA molecules generated in vivo and in vitro are shown in Fig. 1A and B. The results show the presence of the expected dsRNA and additional bands with reduced electrophoretic mobility. IP RP HPLC analysis of the same dsRNAs under non denaturing conditions (50 °C) is also shown in Fig. 1C. The results also show the presence of additional peaks with increased retention times compared to the predominant dsRNA peak. Potential bands with reduced electrophoretic mobility observed on the native agarose gel electrophoresis and increased hydrophobicity observed in IP RP HPLC led us to speculate these dsRNAs are either larger duplex dsRNA species of defined length or potential dsRNA multimers or higher order assemblies. However, in the case of in vitro transcription where dsRNA of defined length is generated by using a DNA template of known length, the production of dsRNA longer than the dsDNA template is not possible. As shown in Fig. 1B, this high molecular weight species was also observed in dsRNA synthesised in vitro from a DNA template of defined length. Therefore, it is unlikely that the bands with reduced mobility are longer dsRNA species of defined size. Rather, the result supports the notion that these are potential dsRNA multimers or higher order assemblies. We have previously shown that under specific non-denaturing IP RP HPLC conditions RNA:RNA interactions are stable and such complexes can be resolved on the basis of differential hydrophobicity in a temperature dependent manner.26
To further characterise the proposed dsRNA multimers observed under native gel electrophoresis and non-denaturing IP RP HPLC, further analysis was performed under denaturing conditions. We have previously demonstrated that the addition of 50% DMSO to dsRNA in conjunction with a short thermal denaturation step is sufficient to denature the dsRNA and prevent re-annealing of the corresponding ssRNAs.22,24 Therefore, this approach was used prior to analysis of the dsRNA using gel electrophoresis (see Fig. 2A). The results show that with addition of the DMSO and a short thermal denaturation step, both the dsRNA and proposed dsRNA multimers are denatured to ssRNA species with similar electrophoretic mobility, indicating that they are the same size. These results demonstrate that the bands with reduced electrophoretic mobility observed under native gel electrophoresis are possibly non-covalent dsRNA multimers. Furthermore, it is evident from the agarose gel analysis that complete denaturation of the dsRNA multimers is only achieved by heating in DMSO and not in DMSO alone. This is consistent with a model of dsRNA aggregation through complex intramolecular and intermolecular base-pair and base-stacking interactions.
To confirm this analysis, further work was performed using IP-RP-HPLC under denaturing conditions (80 °C) see Fig. 2B. The results show that upon thermal denaturation, both the dsRNA and proposed dsRNA multimers (with increased retention time) elute at the same retention time and are therefore the same size, confirming the previous gel electrophoresis analysis. The resulting ssRNA of the same size, as evident by single retention time peak, also confirms that the species with the increased retention time and reduced electrophoretic mobility are not simply larger length ssRNA/dsRNA as this would result in larger length ssRNA and therefore multiple peaks when analysed under denaturing conditions at 80 °C.26
The combined analysis therefore demonstrates that analysis under non-denaturing conditions enables the identification of non-covalent dsRNA multimers. The results demonstrate the ability of dsRNA to form non-covalent multimers following synthesis using in vitro transcription or in vivo in E. coli in conjunction with the extraction methods used in this study.
Further analysis of the non-covalent dsRNA multimers was carried out by first purifying the different RNA species using IP RP HPLC. A number of HPLC fractions were collected and re-injected (see Fig. 3A). The results show the ability to purify the corresponding monomer dsRNA (fraction 1) and dsRNA multimers or higher order assemblies (fractions 2–3), where upon re-injection their retention times remained constant, indicating the different dsRNA species are stable under the conditions employed (see Fig. 3A). Following purification of the dsRNA and dsRNA multimers further analysis of the fractions was performed using gel electrophoresis (see Fig. 3B). The results show that the IP RP HPLC fraction 1 corresponds as expected to the dsRNA and no dsRNA multimers with reduced electrophoretic mobility were observed in this fraction, consistent with previous data. Moreover, the IP-RP-HPLC of fractions 2–3 corresponding to the dsRNA multimers clearly run with reduced electrophoretic mobility under native gel electrophoresis consistent with proposed dsRNA multimers. Further analysis of the IP RP HPLC fractions was performed under denaturing conditions as previously described (see Fig. 3C and D). The analysis of the purified dsRNA multimer fractions 2–3 under denaturing IP-RP-HPLC results in peaks with the same retention time as the dsRNA, confirming the formation of ssRNA species of the same size (see Fig. 3C).
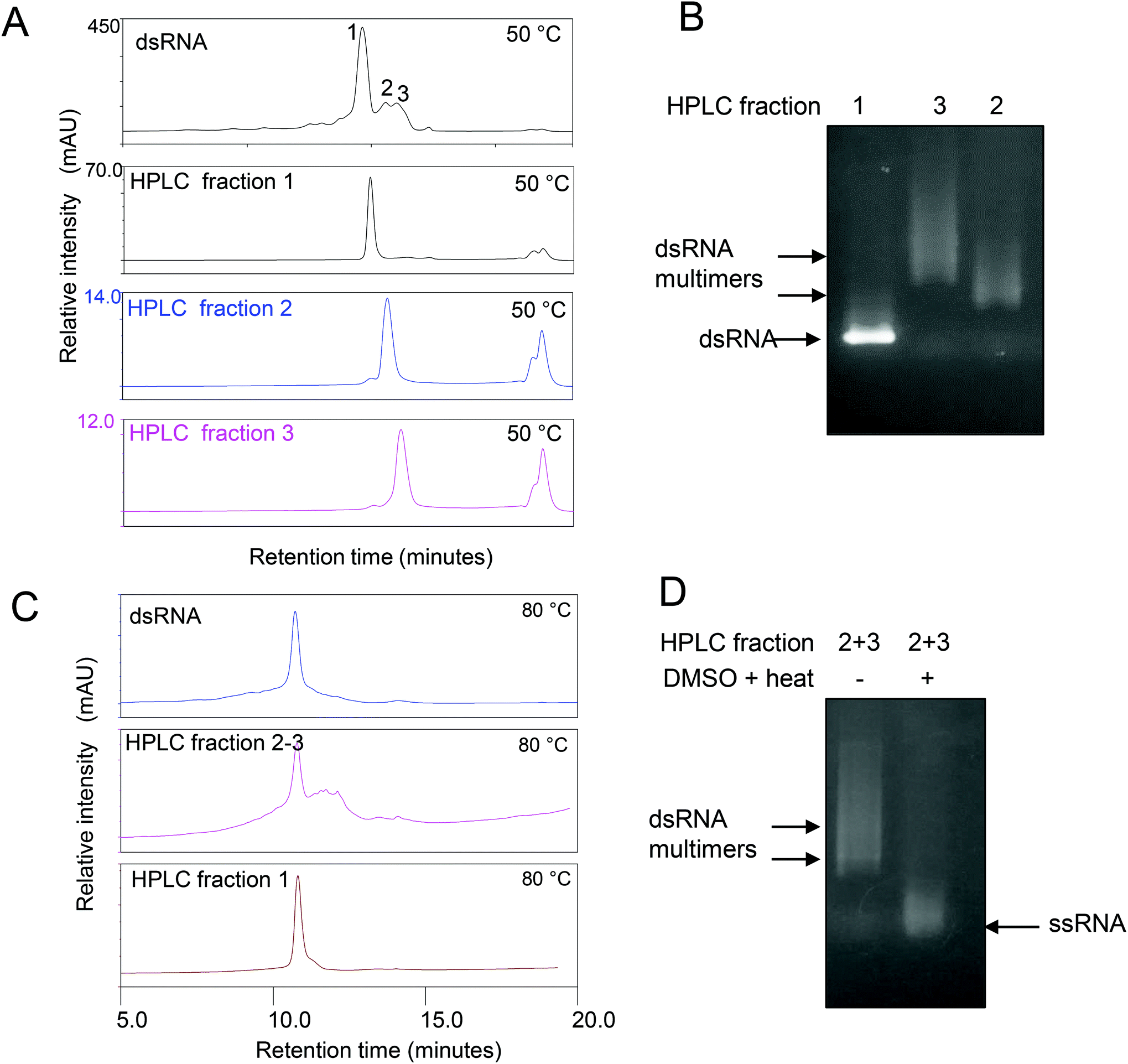 | ||
| Fig. 3 IP RP HPLC fractionation of dsRNA multimers or higher order assemblies. (A) IP RP HPLC chromatogram of the dsRNA synthesized in E. coli. Fractions 1–3 were purified and re-injected prior to HPLC analysis. (B) Non denaturing gel electrophoresis analysis of the corresponding fractions from the IP RP HPLC. The dsRNA multimers or higher order assemblies with reduced electrophoretic mobility are highlighted above the corresponding dsRNA main band. (C) IP RP HPLC chromatograms of the dsRNA fractions 1–3 under denaturing conditions (80 °C). (D) IP RP HPLC fractions were incubated in the presence and absence of 50% DMSO and a short thermal denaturation step prior to gel electrophoresis. | ||
These results demonstrate the presence of structures distinct from the predicted monomeric dsRNA duplex structure. Moreover, for the first time it is shown that purified dsRNA species are a heterogeneous population containing a proportion of proposed multimers or higher order structures. However, the analysis of long dsRNA using gel electrophoresis and IP RP HPLC does not provide direct structural characterisation of the heterogeneous RNA population produced both in vitro and in vivo, therefore limiting the analysis.
3.2. Structural characterisation of purified long dsRNA synthesised in vitro and in vivo
To further characterise the dsRNA and their associated higher order assemblies we used high resolution AFM. Recent advances in instrumentation and availability of small sensitive cantilevers has made it possible to use AFM to image the helical pitch of dsDNA27,28 and dsRNA17 providing structural details at single molecule resolution. In most studies nickel chloride coated mica has been used to immobilise dsDNA and dsRNA to determine their helical pitch. Mica coated with cationic polymers including poly-L-lysine and poly-L-ornithine, are nickel free methods used to immobilise nucleic acids for high-resolution AFM imaging.29,30 In this work, the samples were immobilized on mica coated with poly-L-ornithine and imaged in water (see Materials and Methods). Our initial work focussed on the high resolution AFM imaging of dsDNA (228 bp) and dsRNA (504 bp) both generated in vitro. Small amplitude tapping mode imaging using a high-speed AFM and a short cantilever was gentle enough not to move the double stranded nucleic acids and to image their grooves in a salt free hydrated state (see Fig. 4A and B). In this study for the first time using AFM we were able to see both the major and minor grooves of dsRNA (see Fig. 4A).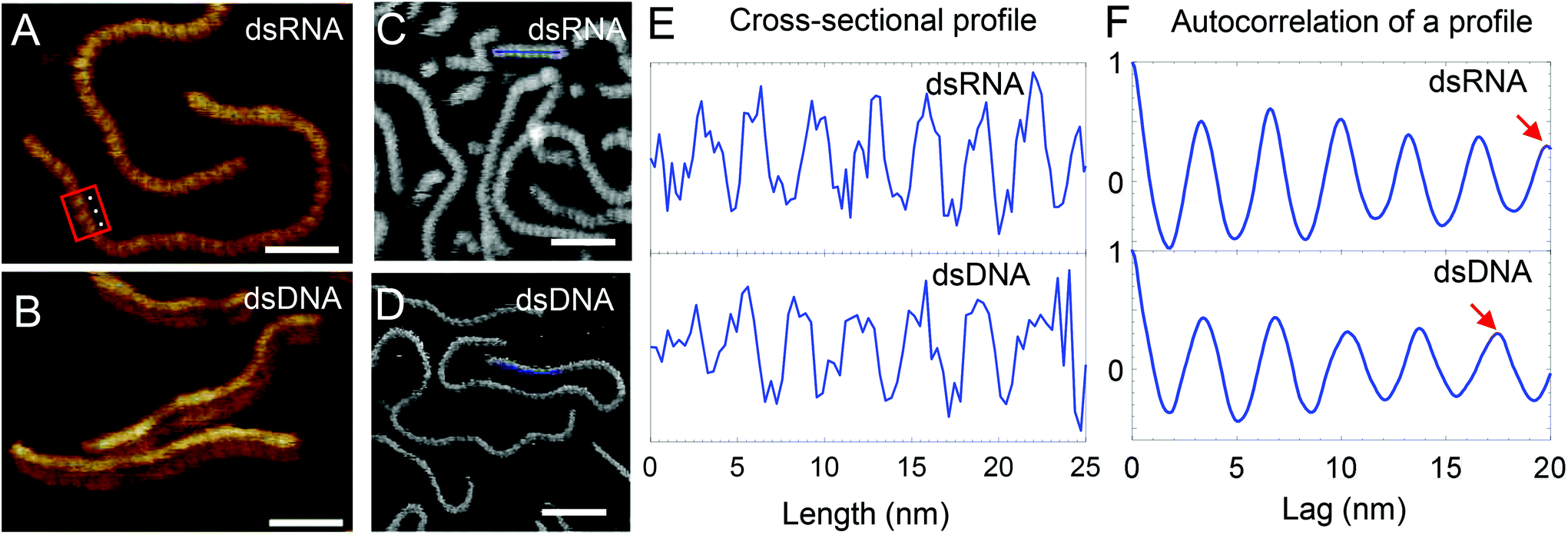 | ||
| Fig. 4 High-resolution AFM images of dsRNA and dsDNA. Red box shows a region where both the major and minor grooves of dsRNA are visible and white dots highlight 3 consecutive grooves. Scale bar in (A) and (B) represents 20 nm while that in (C) and (D) represents 25 nm. Intensity range from dark brown to white for all images is 4 nm. (C/D) Cross-sectional profile (blue line) along AFM images of dsRNA (C) and dsDNA (D) shows their helical pitch in (E). In (E), the profiles were low-pass filtered to remove the low frequency noise. In (F), the positive values of the autocorrelation of a single profile for dsRNA and dsDNA is shown. (F, top) Red arrow points to the 6th peak at 19.8 nm suggesting an average periodicity of 3.3 nm for a dsRNA profile. (F, bottom) Red arrow points to the 5th peak at 17.5 nm suggesting an average periodicity of 3.5 nm for a dsDNA profile (ESI Fig. S1† shows details of periodicity calculations). | ||
Fig. 4E shows cross-sectional profiles of the AFM images of dsRNA and dsDNA that were generated from images shown in Fig. 4C and D and were used to determine the helical pitch of the dsRNA and dsDNA. dsRNA has 8 repetitions of grooves in a 25 nm long profile (see Fig. 4E top). In contrast, dsDNA has 7 grooves in the same length of the profile and this suggests a lower spatial frequency of helical pitch compared to dsRNA (see Fig. 4E bottom). To measure the mean pitch along the nucleic acid polymer, further analysis of the AFM images was performed using autocorrelation of line profiles. From the autocorrelation plot of a line profile, the peak to peak distances were calculated as a measure of periodicity in the profile (see ESI Fig. S1†). In Fig. 4F, the autocorrelation plots show the periodicity for an example profile (Fig. 4F top: dsRNA, Fig. 4F bottom: dsDNA). The peak to peak distances from all the profiles were subsequently used to determine the mean periodicity or the helical pitch. The helical pitch of 3.2 ± 0.2 nm and 3.5 ± 0.1 nm are close to the corresponding theoretical values of 3.1 nm and 3.4 nm for A-form dsRNA and B-form dsDNA (see ESI Table S2,† errors are one standard deviation). A previous study of in air AFM images of dsRNA of known length assuming 11 bases per helical rise corresponds to a pitch of 3.1 nm.16 The pitch of 3.2 nm of dsRNA is far from 3.6 nm, the theoretical pitch of A′-dsRNA (see ESI Table S2†) and hence confirms A-form dsRNA. The difference between the helical pitch from AFM images and theoretical values maybe due to errors introduced due to immobilization, AFM tip–sample interactions, imaging noise, scanner calibration, sample drift and finite pixel size of the AFM images.
3.3. AFM imaging of HPLC purified long dsRNA synthesised in vitro and in vivo
IP RP HPLC was used to purify both the in vitro (504 bp) and in vivo synthesized dsRNA (521 bp) prior to AFM analysis. The IP RP chromatogram of the purified dsRNA is shown in Fig. 5A. Fig. 5C shows cross-sectional profiles of the AFM images of dsRNA that were generated from images shown in Fig. 5B and were used to determine the helical pitch of the dsRNA. From the AFM analysis, it was determined that both the in vitro and in vivo synthesized dsRNA have 9 repetitions of grooves in a 28 nm long profile, demonstrating that they have the same helical pitch and adopt the A-form structure. The data above demonstrates the ability of AFM to provide further direct structural insight of the long dsRNA. In addition to the ability to provide high resolution structural information of the dsRNA, AFM has also previously been utilised to provide accurate size determination of nucleic acids.15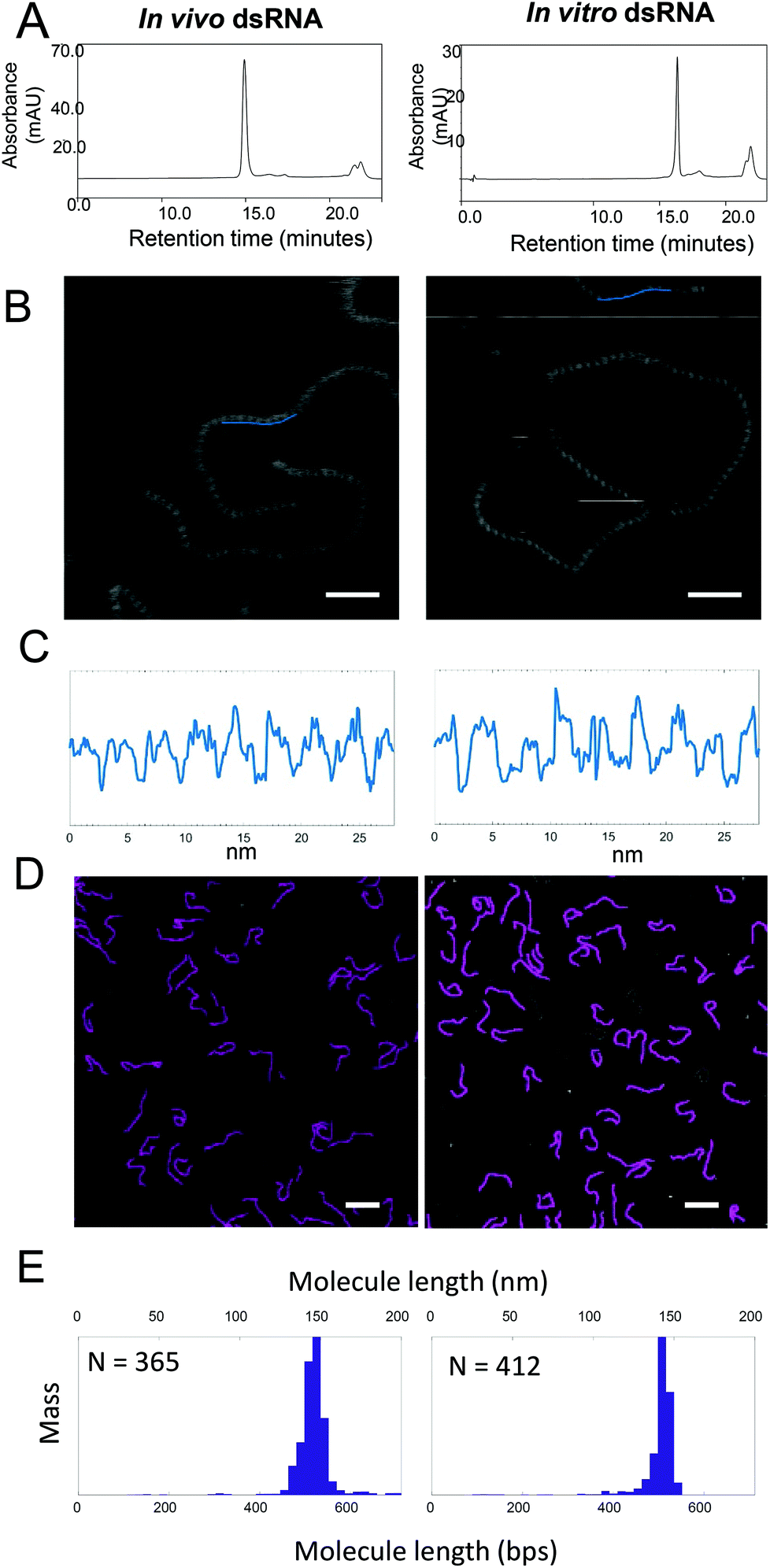 | ||
| Fig. 5 Characterization of purified in vivo and in vitro dsRNA using AFM imaging. (A) IP-RP-HPLC chromatograms of the purified dsRNA synthesized in vivo (521 bp) and in vitro (504 bp). (B) High-resolution AFM images of in vivo and in vitro synthesized dsRNA imaged in water. For (B) the intensity range from dark black to white is 4 nm and scale bar represents 30 nm. (C) Cross-sectional profile of AFM images of in vivo (521 bp) and in vitro (504 bp) synthesized dsRNA. Blue line in (B) shows a 28 nm profile along in vivo and in vitro synthesized dsRNA. In (C) the profiles were low-pass filtered to remove the low frequency noise and the flattened profiles show 9 repeats for both in vivo and in vitro synthesized dsRNA. (D) Representative images of in vivo and in vitro dsRNA in water. Scale bar in (D) represents 100 nm. Individual linear molecules in the AFM images were traced using NeuronJ, an ImageJ plugin (traces are shown in pink). (E) Mass-normalised distribution of the lengths of molecules imaged in water are plotted as histograms. Bin width for histograms are 5 nm. N gives the number of molecules traced. | ||
To determine the size of IP-RP-HPLC purified in vivo and in vitro dsRNA were imaged. Individual linear non-overlapping molecules in the AFM images were subsequently traced using NeuronJ (default parameters), an ImageJ plugin to calculate the lengths of the molecules. Fig. 5D shows representative images of the purified dsRNA generated either in vivo or in vitro. The mass-normalized distribution of the lengths of molecules imaged in water are plotted as histograms shown in Fig. 5E where the lengths in nanometres (nm) were converted to base pairs (bp) by using the theoretical value for rise per base pair of 0.28 nm (see ESI Table S2†). From the size distribution analysis from the image data obtained in water, for in vivo dsRNA, the mean and standard deviation was determined as 523 bp and 18 bp, (which corresponds to 146.5 nm and 5.1 nm) compared to the theoretical predicted size of 521 bp (see ESI Table S3†). For in vitro dsRNA, the mean and standard deviation was determined as 512 bp and 15 bp, (which corresponds to a mean and standard deviation of 143.2 nm and 4.2 nm) compared to the theoretical predicted size of 504 bp. For a control dsDNA, the mean and standard deviation was determined as 220 bp and 17 bp, (which corresponds to 74.8 nm and 5.8 nm) compared to the theoretical predicted size of 228 bp. In addition, further image analysis was also performed in air after drying the samples. The size distribution of the dried sample gave similar results (see ESI Table S4†).
3.4. AFM imaging of dsRNA non-covalent multimers
AFM has previously been used to provide important structural insight into a wide range of different RNA nanostructures in the emerging RNA nanotechnology field.20 Having demonstrated the application of AFM for the high resolution structural confirmation and accurate sizing of the A-form dsRNA generated both in vitro and in vivo, further work was performed to image the proposed non-covalent dsRNA multimers observed under non-denaturing conditions. IP RP HPLC (50 °C) was used to purify the dsRNA and corresponding dsRNA multimers prior to AFM imaging (see Fig. 6). Comparative analysis of the different fractions using AFM clearly demonstrates the formation of different assemblies in each of the fractions (see Fig. 6B). Fraction 1 is consistent with previous analysis demonstrating this sample is A-form dsRNA of approximately 523 bp in length (522 bp in air, see ESI Tables S3 and S4†). The AFM of proposed dsRNA multimers (fractions 2–3) indicates the formation of larger assemblies or structures compared with the AFM analysis of dsRNA fraction 1. Moreover, the AFM structural analysis reveals a more complex potential folding or oligomerisation of the dsRNA. In these images potential overlaps of dsRNA regions or junctions are observed (see Fig. 6B). It is interesting to note that comparative analysis of the dsRNA multimers comparing fractions 2–3 (which demonstrate increased hydrophobicity and overall size of the dsRNA multimers or higher order assemblies) is also reflected in the AFM imaging where increased size of the dsRNA structural assemblies is evident (see Fig. 6A and B). To ensure the images obtained from fractions 2 and 3 are not due to potential overlapping RNA molecules, further dilutions were performed before immobilization on mica coated with poly-L-ornithine. In addition, wide-field views and the size distribution of the multimers or higher order assemblies are shown in ESI Fig. S2.†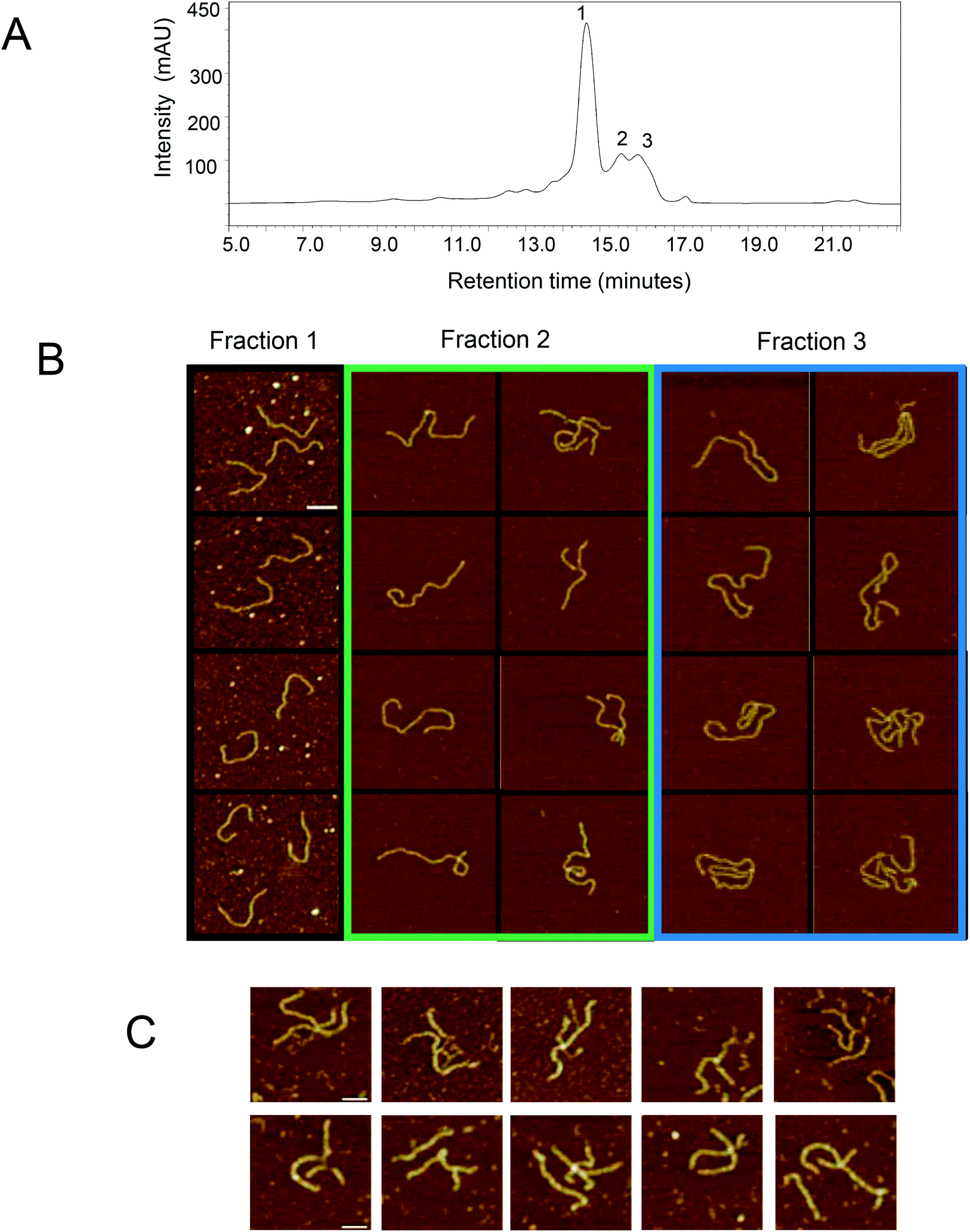 | ||
| Fig. 6 AFM analysis of the non-covalent dsRNA multimers. (A) IP-RP-HPLC chromatogram of 521 bp dsRNA synthesized in E. coli. Fractions 1–3 were collected and analysed using AFM. (B) AFM images of the dsRNA HPLC fractions. All images have the same scale and the scale bar represents 50 nm. Intensity range from dark brown to white for all images is 4 nm (also see ESI Fig. S2†). (C) AFM images of non-covalent dsRNA multimers present in in vitro transcribed 504 bp dsRNA (see Fig. 1C). Representative dsRNA multimers are shown. Images have the same scale and the scale bar represents 30 nm. Intensity range from dark brown to white for all images is 5 nm. | ||
Species with increased hydrophobicity/reduced electrophoretic migration were also observed for in vitro synthesised 504 bp dsRNA, although the intensity of these species was significantly lower compared to dsRNA generated in vivo (see Fig. 1). AFM analysis of the in vitro transcribed dsRNA also revealed the presence of larger assemblies (see Fig. 6C) consistent with AFM analysis of the in vivo generated dsRNA.
4. Conclusions
In this study we have used AFM in conjunction with IP RP HPLC to provide important information regarding both the structural and size heterogeneity of long dsRNA generated both in vitro (using in vitro transcription) and in vivo in bacterial systems. Analytical approaches such as gel electrophoresis, ion-exchange and IP RP HPLC provide no direct structural insight into the long dsRNA. Furthermore, although such approaches enable size determination of the dsRNA, there are a number of caveats associated with accurate size determination using gel electrophoresis and ion exchange chromatography. Curved DNA fragments (including those with AT rich sequences) have been shown to possess reduced mobility during gel electrophoresis.31–33,34 Curved DNA fragments also show retardation during anion exchange HPLC, through the preferential attachment of the curved DNA to the ionic groups of the stationary phase.35 In addition, changes in base composition can affect overall hydrophobicity of the nucleic acids and affect accurate sizing in IP RP HPLC, although this effect is minimal for dsDNA in IP RP HPLC.36 Following IP RP HPLC purification of the dsRNA, AFM analysis in both air and water was performed for >200 individual molecules to determine the length of the corresponding dsRNA. For in vitro dsRNA, a mean of 512 bp ± 15 bp was determined, which corresponds to a mean of 143.2 nm ± 4.2 nm, compared to the theoretical length of 504 bp which corresponds to 141.1 nm.Analysis of dsRNA produced both in vitro and in vivo, using both gel electrophoresis and IP RP HPLC under non-denaturing conditions revealed the presence of potential non-covalent dsRNA multimers or higher order assemblies. Further AFM analysis of the IP RP HPLC purified dsRNA species revealed the formation of larger assemblies or structures compared with the AFM analysis of the monomeric dsRNA, consistent with the formation of dsRNA multimers or higher order assemblies. It is proposed such RNA structures may result from misannealing of the ssRNA species resulting in alternative larger structures/assemblies or multimeric forms of the RNA in contrast to the monomeric duplex dsRNA. Further investigations are required to determine if such species observed under non-denaturing conditions are active in downstream RNAi applications.
In summary, the work presented in this study demonstrates the ability of AFM in conjunction with IP RP HPLC to rapidly assess sample heterogeneity and provide important structural information regarding dsRNA. Furthermore, using these methods enabled us to determine the presence of non-covalent dsRNA multimers or higher order assemblies when analysed under non-denaturing conditions. These approaches increase the repertoire of biophysical and analytical techniques available to study this important biomolecule.
Conflicts of interest
There are no conflicts of interest.Acknowledgements
This work was supported by funding from Syngenta Ltd. MJD acknowledges support from the Biotechnology and Biological Sciences Research Council UK (BBSRC) (BB/M012166/1). JKH and SK acknowledge support from the Engineering and Physical Sciences Research Council UK (EPSRC) (EP/M027430/1).References
- P. A. Sharp, Genes Dev., 1999, 13, 139–141 CrossRef CAS PubMed
.
- R. Katoch, A. Sethi, N. Thakur and L. L. Murdock, Biotechnol. Appl. Biochem., 2013, 171, 847–873 CrossRef CAS PubMed
- A. G. McLoughlin, P. L. Walker, N. Wytinck, D. S. Sullivan, S. Whyard and M. F. Belmonte, Can. J. Plant Pathol., 2018, 40, 1–11 CrossRef
- C. Ratzka, R. Gross and H. Feldhaar, Insectes Soc., 2013, 60, 475–484 CrossRef
- J.-Q. Zhu, S. Liu, Y. Ma, J.-Q. Zhang, H.-S. Qi, Z.-J. Wei, Q. Yao, W.-Q. Zhang and S. Li, PLoS One, 2012, 7, e38572 CrossRef CAS PubMed
- M. L. Taracena, P. L. Oliveira, O. Almendares, C. Umaña, C. Lowenberger, E. M. Dotson, G. O. Paiva-Silva and P. M. Pennington, PLoS Neglected Trop. Dis., 2015, 9, e0003358 CrossRef PubMed
- R. Bolognesi, P. Ramaseshadri, J. Anderson, P. Bachman, W. Clinton, R. Flannagan, O. Ilagan, C. Lawrence, S. Levine, W. Moar, G. Mueller, J. G. Tan, J. Uffman, E. Wiggins, G. Heck and G. Segers, PLoS One, 2012, 7, e47534 CrossRef CAS PubMed
- S. C. Miller, K. Miyata, S. J. Brown and Y. Tomoyasu, PLoS One, 2012, 7, e47431 CrossRef CAS PubMed
- S. Ivashuta, Y. Zhang, B. E. Wiggins, P. Ramaseshadri, G. C. Segers, S. Johnson, S. E. Meyer, R. A. Kerstetter, B. C. McNulty and R. Bolognesi, RNA, 2015, 5, 840–850 CrossRef PubMed
- M. Yaneva, T. Kowalewski and M. R. Lieber, EMBO J., 1997, 16, 5098–5112 CrossRef CAS PubMed
- H. G. Hansma, M. Bezanilla, F. Zenhausern, M. Adrian and R. L. Sinsheimer, Nucleic Acids Res., 1993, 21, 505–512 CrossRef CAS PubMed
- J. Mou, D. M. Czajkowsky, Y. Zhang and Z. Shao, FEBS Lett., 1995, 371, 279–282 CrossRef CAS PubMed
- H. Wang, I. B. Dodd, D. D. Dunlap, K. E. Shearwin and L. Finzi, Nucleic Acids Res., 2013, 41, 5746–5756 CrossRef CAS PubMed
-
P. Schön, Atomic force microscopy of RNA: State of the art and recent advancements, in Semin. Cell Dev. Biol., Elsevier, 2017 Search PubMed
- Y. L. Lyubchenko, B. L. Jacobs and S. M. Lindsay, Nucleic Acids Res., 1992, 20, 3983–3986 CrossRef CAS PubMed
- E. Herrero-Galán, M. E. Fuentes-Perez, C. Carrasco, J. M. Valpuesta, J. L. Carrascosa, F. Moreno-Herrero and J. R. Arias-Gonzalez, J. Am. Chem. Soc., 2012, 135, 122–131 CrossRef PubMed
- P. Ares, M. E. Fuentes-Perez, E. Herrero-Galán, J. M. Valpuesta, A. Gil, J. Gomez-Herrero and F. Moreno-Herrero, Nanoscale, 2016, 8, 11818–11826 RSC
- S. Petkovic, S. Badelt, S. Block, C. Flamm, M. Delcea, I. Hofacker and S. Müller, RNA, 2015, 21, 1249–1260 CrossRef CAS PubMed
- J. Pallesen, M. Dong, F. Besenbacher and J. Kjems, FEBS J., 2009, 276, 4223–4232 CrossRef CAS PubMed
- P. Guo, Nat. Nanotechnol., 2010, 5, 833–842 CrossRef CAS PubMed
- L. Timmons, D. L. Court and A. Fire, Gene, 2001, 263, 103–112 CrossRef CAS PubMed
- A. O. Nwokeoji, A.-W. Kung, P. M. Kilby, D. E. Portwood and M. J. Dickman, J. Chromatogr. A, 2017, 1484, 14–25 CrossRef CAS PubMed
- A. O. Nwokeoji, P. M. Kilby, D. E. Portwood and M. J. Dickman, Anal. Biochem., 2016, 512, 36–46 CrossRef CAS PubMed
- A. O. Nwokeoji, P. M. Kilby, D. E. Portwood and M. J. Dickman, Anal. Chem., 2017, 89, 13567–13574 CrossRef CAS PubMed
- S. Kumar, M. L. Cartron, N. Mullin, P. Qian, G. J. Leggett, C. N. Hunter and J. K. Hobbs, ACS Nano, 2016, 11, 126–133 CrossRef PubMed
- S. P. Waghmare, P. Pousinis, D. P. Hornby and M. J. Dickman, J. Chromatogr. A, 2009, 1216, 1377–1382 CrossRef CAS PubMed
- C. Leung, A. Bestembayeva, R. Thorogate, J. Stinson, A. Pyne, C. Marcovich, J. Yang, U. Drechsler, M. Despont, T. Jankowski, M. Tschöpe and B. W. Hoogenboom, Nano Lett., 2012, 12, 3846–3850 CrossRef CAS PubMed
- A. Pyne, R. Thompson, C. Leung, D. Roy and B. W. Hoogenboom, Small, 2014, 10, 3257–3261 CrossRef CAS PubMed
- B. Klejevskaja, A. L. Pyne, M. Reynolds, A. Shivalingam, R. Thorogate, B. W. Hoogenboom, L. Ying and R. Vilar, ChemComm, 2016, 52, 12454–12457 RSC
-
A. L. Pyne and B. W. Hoogenboom, in Chromosome Architecture, Springer, 2016, pp. 47–60 Search PubMed
- J. C. Marini, S. D. Levene, D. M. Crothers and P. T. Englund, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 7664–7668 CrossRef CAS PubMed
- N. C. Stellwagen, Biochemistry, 1983, 22, 6180–6185 CrossRef CAS PubMed
- H.-M. Wu and D. M. Crothers, Nature, 1984, 308, 509–513 CrossRef CAS PubMed
- O. J. Lumpkin, P. Déjardin and B. H. Zimm, Biopolymers, 1985, 24, 1573–1593 CrossRef CAS PubMed
- F. Fack and V. Sarantoglou, Nucleic Acids Res., 1991, 19, 4181–4188 CrossRef CAS PubMed
- M. J. Dickman, J. Chromatogr. A, 2005, 1076, 83–89 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9an00954j |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2019 |