
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Separation of isomeric glycans by ion mobility spectrometry – the impact of fluorescent labelling†
Christian
Manz
 ab,
Márkó
Grabarics
ab,
Friederike
Hoberg
b,
Michele
Pugini
ab,
Alexandra
Stuckmann
a,
Weston B.
Struwe
*c and
Kevin
Pagel
*ab
ab,
Márkó
Grabarics
ab,
Friederike
Hoberg
b,
Michele
Pugini
ab,
Alexandra
Stuckmann
a,
Weston B.
Struwe
*c and
Kevin
Pagel
*ab
aInstitute of Chemistry and Biochemistry, Freie Universität Berlin, Takustrasse 3, 14195 Berlin, Germany. E-mail: kevin.pagel@fu-berlin.de
bFritz Haber Institute of the Max Planck Society, Department of Molecular Physics, Faradayweg 4-6, 14195 Berlin, Germany
cOxford Glycobiology Institute, Department of Biochemistry, University of Oxford, Oxford OX1 3QU, UK. E-mail: weston.struwe@chem.ox.ac.uk
First published on 26th July 2019
Abstract
The analysis of complex oligosaccharides is traditionally based on multidimensional workflows where liquid chromatography is coupled to tandem mass spectrometry (LC-MS/MS). Due to the presence of multiple isomers, which cannot be distinguished easily using tandem MS, a detailed structural elucidation is still challenging in many cases. Recently, ion mobility spectrometry (IMS) showed great potential as an additional structural parameter in glycan analysis. While the time-scale of the IMS separation is fully compatible to that of LC-MS-based workflows, there are very few reports in which both techniques have been directly coupled for glycan analysis. As a result, there is little knowledge on how the derivatization with fluorescent labels as common in glycan LC-MS affects the mobility and, as a result, the selectivity of IMS separations. Here, we address this problem by systematically analyzing six isomeric glycans derivatized with the most common fluorescent tags using ion mobility spectrometry. We report >150 collision cross-sections (CCS) acquired in positive and negative ion mode and compare the quality of the separation for each derivatization strategy. Our results show that isomer separation strongly depends on the chosen label, as well as on the type of adduct ion. In some cases, fluorescent labels significantly enhance peak-to-peak resolution which can help to distinguish isomeric species.
Introduction
Carbohydrates are the most abundant biopolymers on Earth.1 The majority are large and regular polysaccharides, which often serve as structural scaffolds such as in cellulose, or energy sources in nutrition such as in starch. However, many biological functions are performed by smaller, more complex oligosaccharides, often referred to as glycans. They for example play a role in the prebiotic nutrition of infants2 and regulate immune responses.3,4 In contrast to oligonucleotides and peptides, the biosynthesis of oligosaccharides is not template-driven, leading to structures that are highly diverse with a complex branching pattern, regio- and stereochemistry. Mass spectrometry (MS) analysis is in most cases not sufficient to unambiguously identify the structure of glycans, since multiple isomers often coexist. As a result of this complexity, an in-depth glycan analysis still presents a major analytical challenge.5A common way to resolve and separate glycan isomers is liquid chromatography (LC). Reversed phase (RP) chromatography, which is commonly used for protein analysis, often struggles with glycans due to their inherently high polarity. Instead, other stationary phases such as hydrophilic interaction chromatography (HILIC) and porous graphitic carbon (PGC) are often applied as an alternative, powerful way to separate glycan isomers.6 However, as glycans naturally do not contain chromophores or fluorophores, it is often necessary to derivatize them with fluorescent labels to facilitate a sufficient detection and enable quantification.7
Another emerging and promising technique capable of separating glycan isomers is ion mobility spectrometry (IMS).5,8,9 Here, ions travel through a drift cell filled with an inert buffer gas under the influence of a weak electric field and undergo low-energy collisions with the buffer gas. Compact ions collide less frequently with the buffer gas than more extended ions, which leads to a separation based on size, shape and charge. This enables the separation of isomeric species as shown for small molecules,10 oligosaccharides as well as for glycoconjugates.11,12 In addition, the resulting drift times can be converted into the rotationally averaged collision cross-section (CCS). When measured under controlled conditions, CCSs can be universally compared, which enables an efficient incorporation into databases to allow for structural elucidation.13–17 While over the last several years both LC and IMS, showed their individual capabilities to resolve glycan isomers, very few attempts have been made to combine both methods into a consistent LC-IM-MS workflow for glycan analysis.18 Importantly, the impact of derivatization, in particular with fluorescence labels, on the mobility separation of isomeric glycans is poorly understood. To close this gap, we present a systematical analysis using a set of isomeric glycans derivatized with different common glycan fluorophores as well as native and reduced species. Our data indicate that labelling can significantly affect the ability to separate individual glycan isomers via IM-MS. Depending on the label, this can diminish or improve selectivity, and therefore, labels should be specifically selected for a given glycan analysis.
Experimental
Sample preparation
All labelling reagents and solvents were purchased from Sigma Aldrich (St Louis, USA) and used without further purification. Synthetically derived Lewis oligosaccharides were purchased from Dextra Laboratories Ltd (Reading, UK). Prior to analysis, the Lewis antigens were diluted to 1 mM stock solution in HPLC grade water. The stock solution was divided into 10 μL (10 nmol) aliquots and freeze dried. Dried Lewis antigens were labelled with 2-aminobenzoic acid (2-AA), 2-aminobenzamide (2-AB), 4-amino-N-[2-(diethylamino)ethyl]benzamide (procainamide, ProA) via reductive amination.19 Removal of excess label was performed using paper chromatography.20 Alditols were synthezised via reduction with sodium borohydride.21 The reduced, as well as 2-AB, 2-AA and ProA labelled glycans were further purified using HyperSep Hypercarb SPE cartridges (ThermoFisher Scientific, Waltham, Massachusetts, US) according to manufacturer's instructions. Afterwards, the purified glycans were freeze dried and redissolved in HPLC grade water to yield a ∼100 μM stock solution.Ion mobility-mass spectrometry
Linear drift tube (DT) IM-MS measurements were performed on a modified Synapt G2-S HDMS instrument (Waters Corporation, Manchester, UK), described in detail elsewhere.22 Measurements were performed in positive and negative ion mode with platinum/palladium (Pt/Pd, 80/20) coated borosilicate capillaries prepared in-house. Prior to measurements, each sample was diluted from stock solution with methanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water (1:1) to result in a final concentration of 10 μM. Salt solutions were generated by adding a 10 mg mL−1 aqueous stock solution of KCl/LiCl/NaCl to the labelled glycan solution to result in a 1:5 ratio (salt:glycan).
:water (1:1) to result in a final concentration of 10 μM. Salt solutions were generated by adding a 10 mg mL−1 aqueous stock solution of KCl/LiCl/NaCl to the labelled glycan solution to result in a 1:5 ratio (salt:glycan).
For nano-electrospray ionization (nano-ESI) typically 5 μL of sample was loaded to a capillary and electrosprayed by applying a capillary voltage of 0.6–1.1 kV. Typical parameters in positive ion mode were: 60 V sampling cone voltage, 1 V source offset voltage, 30 °C source temperature, 0 V trap CE (MS) up to 30 V trap CE (MSMS), 2 V transfer CE, 3 mL min−1 trap gas flow. Ion mobility parameters were: 2.2 Torr helium IMS gas, 27–30 °C IMS temperature, 5.0 V trap DC entrance voltage, 5.0 V trap DC bias voltage, −10.0 V trap DC voltage, 2.0 V trap DC exit voltage, −25.0 V IMS DC entrance voltage, 50–180 V helium cell DC voltage, −40.0 V helium exit voltage, 50–150 V IMS bias voltage, 0 V IMS DC exit voltage, 5.0 V transfer DC entrance voltage, 15.0 V transfer DC exit voltage, 150 m s−1 trap wave velocity, 1.0 V trap wave height voltage, 200 m s−1 transfer wave velocity, 5.0 V transfer wave height voltage.
In negative ion mode typical parameters were: 90 V sampling cone voltage, 10 V source offset voltage, 30 °C source temperature, 0 V trap CE (MS) up to 30 V trap CE (MSMS), 2 V transfer CE, 3 mL min−1 trap gas flow. Ion mobility parameters were: 2.2 Torr helium IMS gas, 27–30 °C IMS temperature, 1.0 V trap DC entrance voltage, 2.0 V trap DC bias voltage, −1.0 V trap DC voltage, 1.5 V trap DC exit voltage, −25.0 V IMS DC entrance voltage, 50–150 V helium cell DC voltage, −40.0 V helium exit voltage, 50–150 V IMS bias voltage, 0 V IMS DC exit voltage, 5.0 V transfer DC entrance voltage, 15.0 V transfer DC exit voltage, 200 m s−1 trap wave velocity, 10.0 V trap wave height voltage, 250 m s−1 transfer wave velocity, 3.0 V transfer wave height voltage. The resulting drift times were converted to rotationally-averaged collision cross-sections (CCS) using the Mason–Schamp equation.23
Results and discussion
ABO and Lewis blood group system
A common and widely studied set of isomeric glycans are the epitopes of two different blood group systems: the Lewis antigens (Le) and the ABO blood group system (H antigen, commonly referred to as BG H). These structures are typically found as features on larger glycoconjugates such a N- and O-linked glycans or on human milk oligosaccharides (HMO).24 The two main types of Lewis antigens are the tetrasaccharide motifs LeY and LeB, which feature a common monosaccharide composition and are displayed in Fig. 1. Type 1 antigens (LeB series) consist of galactose (Gal) β-(1→3) linked to N-acetylglucosamine (GlcNAc) core, whereas type 2 antigens (LeY-series) contain a β-(1→4) linked core. Both antigens are functionalized by two differently attached fucose (Fuc) units. The loss of one fucose unit leads to the regioisomeric trisaccharides LeX and BG H2 (LeY-series) or LeA and BG H1 (LeB-series). In larger glycan structures, epitopes are formed by terminal fucosylation. Each of the resulting epitopes has specific functional properties and alteration is often associated with pathological processes, including cancer progression and atherosclerosis.26,27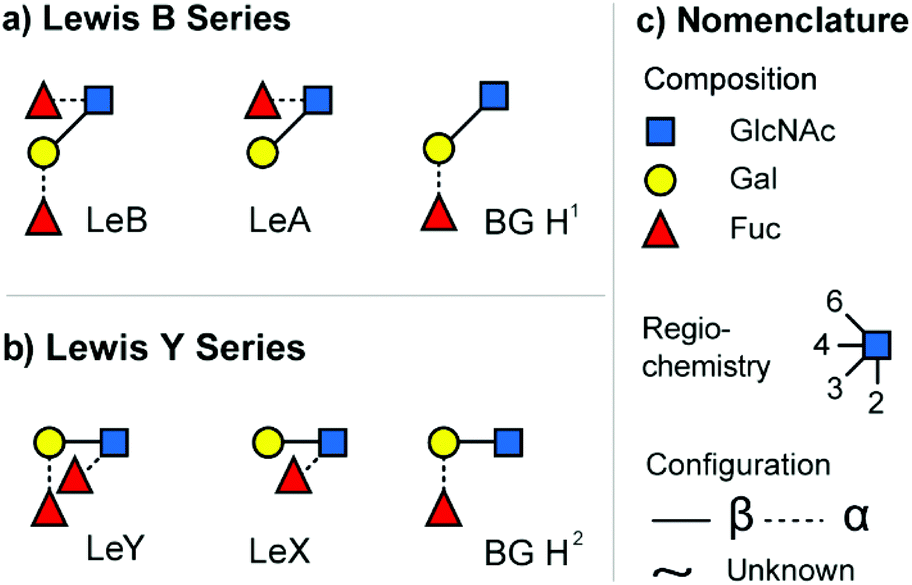 | ||
| Fig. 1 The investigated set of isomeric blood group epitopes. (a) Tetrasaccharide Lewis B (LeB) and the corresponding trisaccharide fragments/motifs Lewis A (LeA) and blood group H1 (BG H1). (b) Tetrasaccharide Lewis Y (LeY) and the corresponding trisaccharide fragments/motifs Lewis X (LeX) and blood group H2 (BG H2). (c) Glycan structures are depicted using the SNFG nomenclature.25 | ||
Recently, we investigated these isomeric tri- and tetrasaccharides in an underivatized form using IM-MS and showed that fragment CCS can be used as fingerprints to systematically differentiate between the epitopes.28 The intact tetrasaccharide precursors exhibit very similar drift times and CCS, which makes it difficult to distinguish them using IMS. However, fragmentation of the tetrasaccharide precursors with collision induced dissociation (CID) yields trisaccharide fragments that can be used to identify specific terminal fucose motifs. Some of those isomeric fragments such as LeX and BG H2 can be readily distinguished by IMS, while LeA and BG H1 are difficult to differentiate in underivatized form. In the present study, we focus on the IMS separation of derivatized forms of these epitopes.
Labelling of glycans
Common glycan derivatization strategies, not only for the ABO and Lewis system, include permethylation, reduction, and various reducing end modifications via reductive amination.19 Reducing end modifications have been studied extensively, namely the influence of fluorescent labels on retention in various chromatographic modes,29–31 ionization efficiency in electrospray ionization (ESI),32 fragmentation patterns in MS33,34 and on rearrangement reactions of glycan ions.35,36 On the other hand, very little is known about their influence on ion mobility separations. In the present study, we focus on the four most common reducing end modifications to study their influence on IMS separation (Fig. 2). Due to the high labelling efficiency of reductive amination and the stability of the resulting labelled glycans, the fluorophores 2-aminobenzoic acid (2-AA) and 2-aminobenzamide (2-AB) are currently the most commonly used labels. They are readily available and known for their sensitivity in fluorescence detection. Procainamide (ProA) is a beneficial fluorophore used for coupling LC with MS because its tertiary amine moiety significantly enhances ionization efficiency in matrix-assisted laser desorption/ionization (MALDI) and ESI.37,38 Since all three labels have a hydrophobic character, they increase retention of the inherently polar glycans in reversed-phase separations. As only one label is incorporated per individual glycan, derivatization with 2-AA, 2-AB or ProA furthermore enables a simple quantification.39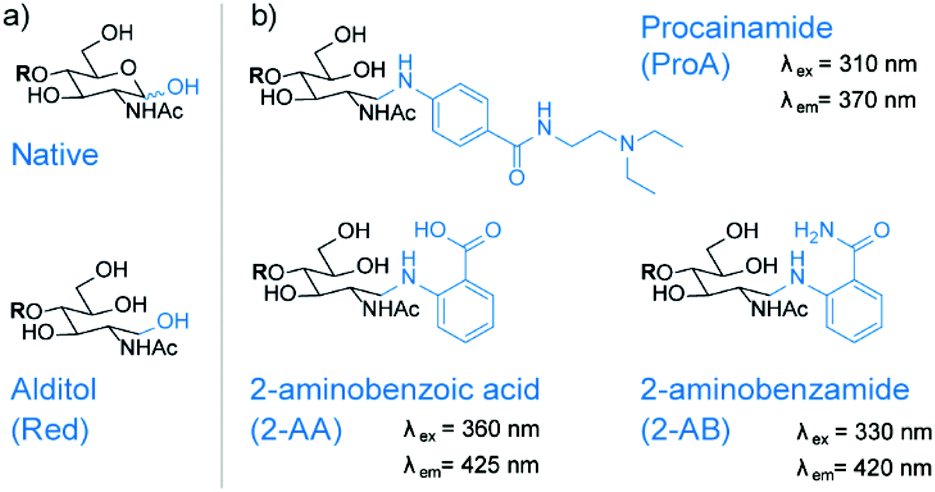 | ||
| Fig. 2 Reducing end modifications investigated in this study. (a) Native glycan and the reduced alditol structure without chromophore at reducing end. (b) Glycan derivatization with chromophore such as procainamide (ProA), 2-aminobenzoic acid (2-AA) and 2-aminobenzamide (2-AB) is a common strategy to increase sensitivity and enable quantification. For some labels there are additional benefits such as an improved ionization efficiency in ESI or an improved selectivity in HPLC. | ||
After reductive amination with fluorescent labels such as 2-AB, 2-AA or ProA, the reducing end monosaccharide will exhibit an open ring structure. To study the influence of this ring opening, we reduced glycans to open-ring alditols (Red) to compare them with the predominantly closed-ring native structures. The reduction of glycans often precedes permethylation, but alditols themselves are also often used as standalone modification for various stationary phases.39
Ion mobility separation of labelled glycans
In order to address the impact of labelling on the CCS of glycans, the set of blood group antigens shown in Fig. 1 was subjected to reducing end modifications displayed in Fig. 2. For all modified species the drift times in helium drift gas were measured and CCSs calculated (DTCCSHe). Measured arrival time distributions (ATDs) of the two fucosylated trisaccharide isomer pairs LeX/BG H2 and LeA/BG H1 as sodiated species with different reducing end modifications are shown in Fig. 3. All ATDs were measured under the very same instrumental conditions (such as pressure, drift voltages and drift gas).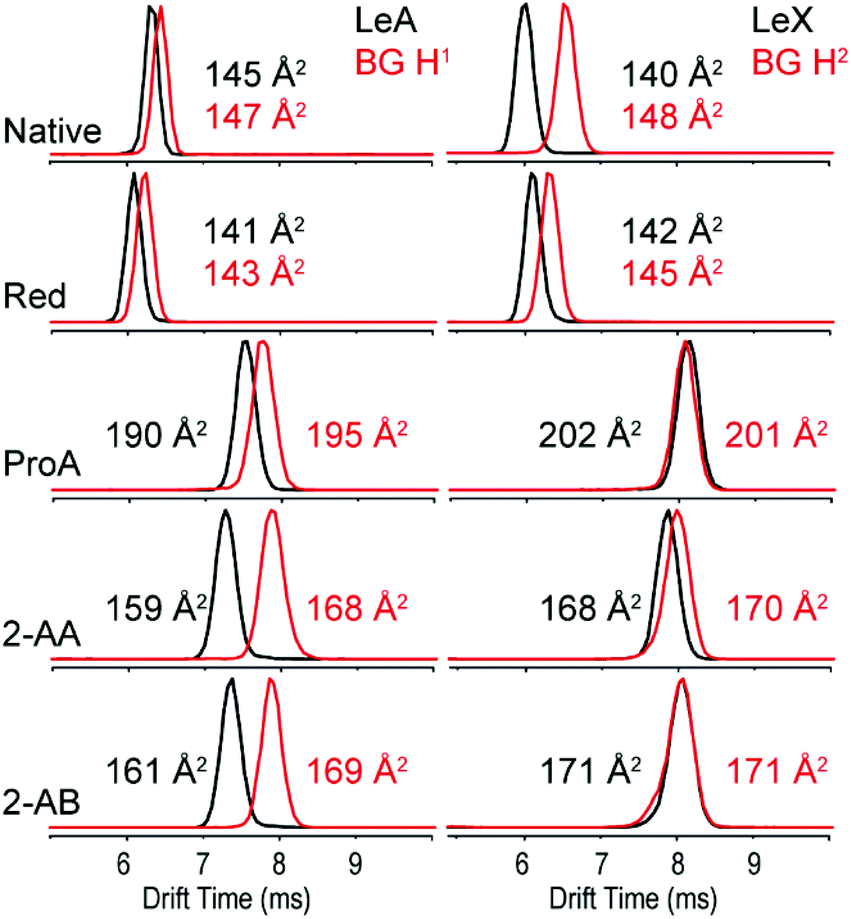 | ||
| Fig. 3 ATDs and DTCCSHe of the isomeric blood group epitopes LeA vs. BG H1 (left panel) and LeX vs. BG H2 (right panel) as sodium adducts in He drift gas. | ||
As native glycans, LeA and BG H1 show minor isomer separation compared to LeX and BG H2, which are almost baseline separated. Compared to the native closed-ring structure, the ring opening during reduction to alditols does not seem to have a significant impact on the separation of LeA and BG H1. In contrast, the drift-time difference of LeX and BG H2 decreases significantly after reduction. This effect is further amplified after introduction of the chromophore labels ProA, 2-AA and 2-AB. 2-AA and 2-AB labelled glycans show the largest isomer separation for the LeA and BG H1 isomers, while LeX and BG H2 are basically indistinguishable.
Similarly, all native and derivatized isomers were measured as alkali metal adducts, which are known to significantly alter isomer separation in IMS.40 Negatively charged adducts such as chloride and nitrate complexes predominantly lead to the formation of deprotonated ions, which are therefore the only ions with negative polarity studied here.41,42 In Table 1, the CCSs of protonated and deprotonated glycans, as well as for three commonly observed typical alkali adducts (Li+, Na+, K+) measured in helium (DTCCSHe) are shown. The upper part of Table 1 shows the CCSs of all derivatives and metal adducts of the LeB series, while the lower part shows the CCSs of all modifications of the LeY series. The exact masses of all labelled glycans are shown in Table S1.†
| DTCCSHe (Å2) | Type | Native | Red | 2-AB | 2-AA | ProA |
|---|---|---|---|---|---|---|
| LeB | [M + H]+ | 167 | 167 | 189 | 189 | 209 |
| [M + Li]+ | 165 | 159 | 179 | 178 | 209 | |
| [M + Na]+ | 166 | 160 | 180 | 178 | 210 | |
| [M + K]+ | 166 | 164 | 182 | 180 | 214 | |
| [M − H]− | 165 | 161 | 184 | 179 | 218 | |
| LeA | [M + H]+ | 144 | 144 | 168 | 167 | 183 |
| [M + Li]+ | 143 | 138 | 159 | 158 | 188 | |
| [M + Na]+ | 145 | 141 | 161 | 159 | 190 | |
| [M + K]+ | 145 | 146 | 163 | 163 | 194 | |
| [M − H]− | 143 | 141 | 162 | 160 | 190 | |
| BG H1 | [M + H]+ | 144 | 144 | 170 | 168 | 188 |
| [M + Li]+ | 146 | 142 | 167 | 167 | 195 | |
| [M + Na]+ | 147 | 143 | 169 | 168 | 195 | |
| [M + K]+ | 145 | 146 | 169 | 170 | 193 | |
| [M − H]− | 143 | 143 | 163 | 157 | 192 | |
| LeY | [M + H]+ | 171 | 169 | 190 | 191 | 209 |
| [M + Li]+ | 163 | 163 | 190 | 190 | 219 | |
| [M + Na]+ | 164 | 164 | 192 | 191 | 218 | |
| [M + K]+ | 165 | 166 | 192 | 191 | 217 | |
| [M − H]− | 167 | 164 | 185 | 183 | 213 | |
| LeX | [M + H]+ | 144 | 145 | 169 | 170 | 188 |
| [M + Li]+ | 138 | 141 | 169 | 165 | 202 | |
| [M + Na]+ | 140 | 142 | 171 | 168 | 202 | |
| [M + K]+ | 141 | 144 | 171 | 169 | 198 | |
| [M − H]− | 146 | 141 | 161 | 159 | 189 | |
| BG H2 | [M + H]+ | 144 | 145 | 172 | 166 | 185 |
| [M + Li]+ | 148 | 145 | 170 | 169 | 201 | |
| [M + Na]+ | 148 | 146 | 171 | 170 | 201 | |
| [M + K]+ | 149 | 148 | 172 | 172 | 197 | |
| [M − H]− | 144 | 139 | 163 | 155 | 190 | |
Generally, there is a clear trend of increasing CCS from the native glycans up to the ProA-labelled glycans. The CCSs for all species is growing proportionally to the size of the added fluorescent label, and is therefore correlated to the increase in molecular mass.40 A similar trend is observed with the addition of alkali metals, which generally lead to larger CCSs in the order of H+ < Li+ < Na+ < K+. Deprotonated species on the other hand behave similarly counterparts. However, there are some exceptions to this behaviour. Especially alditols (Red) seem to have their largest CCS when protonated or adducted with potassium, while sodiated and lithiated species show significantly smaller CCSs. Another example are glycans labelled with 2-AA, whose protonated species show larger CCSs than metal adducted species, which indicates a compaction of the gas-phase structure with the addition of small alkali metal ions. This behaviour is a result of the structure of the oligosaccharide-metal complex, which is dictated by the solvation of the metal cation.
Comparison of isomeric labelled glycan isomers
As shown above, the CCS of each species strongly depends on the modification of the reducing end and the type of adduct. The type of label will therefore also affect the ability to separate isomers. Based on the CCS for each individual species it is difficult to rank the quality of each individual separation. A highly useful index for evaluating this performance is the peak-to-peak resolution (Rs). It serves as a quantitative measure of the extent to which a pair of peaks is separated. The definition of Rs – universally accepted in column chromatography, ion mobility spectrometry, etc. – is given by the following equation: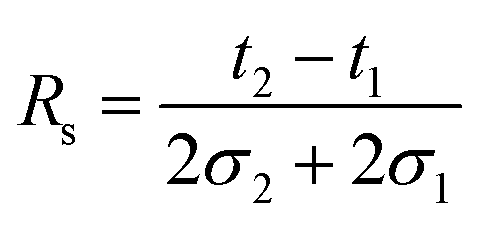 | (1) |
Here, 2σ is the temporal peak width measured between the two inflection points of a peak of Gaussian profile and t the drift time. In case of separating isomers of the same charge state by linear drift tube ion mobility spectrometry (DTIMS), eqn (1) can be rewritten in the following form:
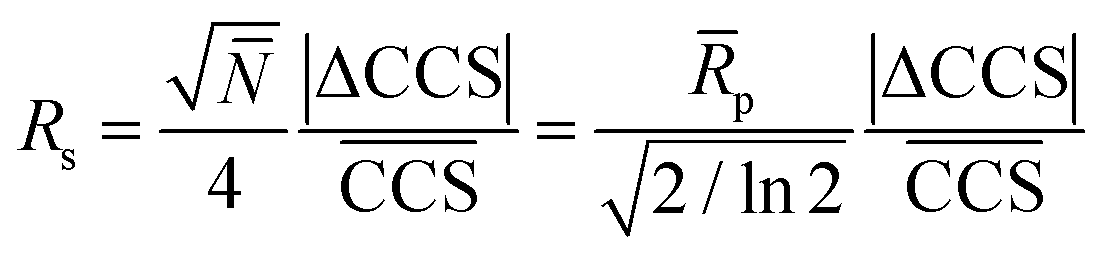 | (2) |
In eqn (2)N and ![[R with combining macron]](https://www.rsc.org/images/entities/i_char_0052_0304.gif) p are the average plate number and average resolving power, respectively (the latter one should not be confused with peak-to-peak resolution). The relation between the two figures of merit is given by definition as
p are the average plate number and average resolving power, respectively (the latter one should not be confused with peak-to-peak resolution). The relation between the two figures of merit is given by definition as 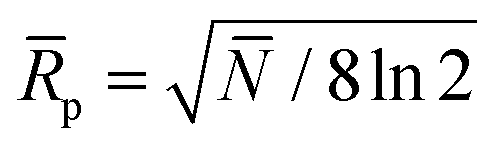 , the origin of the constant in the formula being the difference between the standard deviation and the full-width-at-half-maximum (FWHM) of Gaussian distributions. The fraction
, the origin of the constant in the formula being the difference between the standard deviation and the full-width-at-half-maximum (FWHM) of Gaussian distributions. The fraction 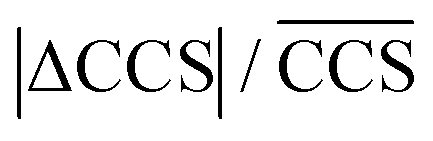 is the relative difference between the collision cross sections of the ions that are to be separated, i.e. it is a measure of selectivity. In following, we used this term to describe the difference in CCS between the tetrasaccharide isomer pair LeY/LeB as well as the trisaccharide isomer pairs LeX/BG H2 and LeA/BG H1 and visualize this difference as heat map in Table 2.
is the relative difference between the collision cross sections of the ions that are to be separated, i.e. it is a measure of selectivity. In following, we used this term to describe the difference in CCS between the tetrasaccharide isomer pair LeY/LeB as well as the trisaccharide isomer pairs LeX/BG H2 and LeA/BG H1 and visualize this difference as heat map in Table 2.
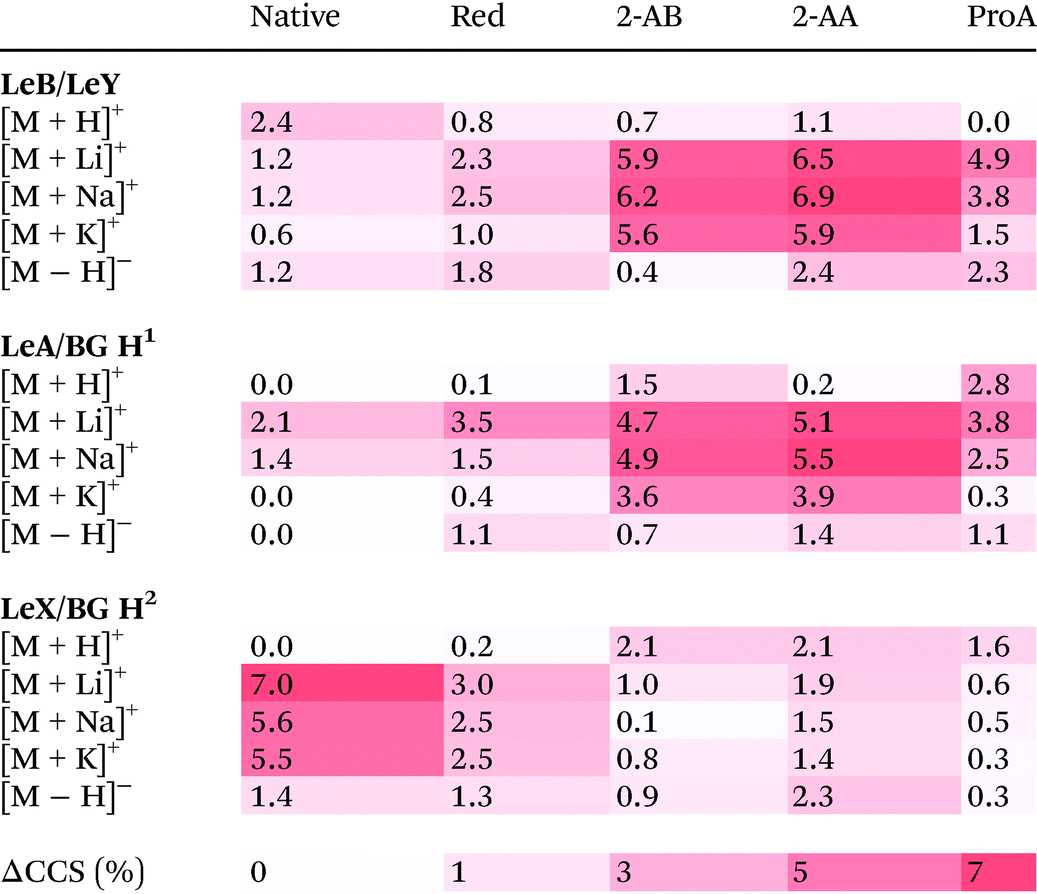
|
The upper part of Table 2 shows the isomer separation for the tetrasaccharides LeY/LeB. Here two general trends are observed: (1) isomer separation is increased when a fluorescent label is introduced and (2) isomer separation is improved upon adduct formation with alkali metal adducts. There are, however, significant differences between each individual modification. While native tetrasaccharides only show up to 2.4% CCS difference, 2-AA and 2-AB labelled species separate much better with a difference of up to 6.9% for sodium adducts. ProA-labelled isomers, on the other hand, are separated as lithiated species with a difference of 4.9%. Thus, specific labels can increase isomer separation, which in some cases makes them beneficial for IMS separation.
A very similar behaviour of improved separation is observed for the LeB submotifs LeA vs. BG H1. Native structures of these isomers do practically not separate in IMS independent of the charge carrier; only lithiated and sodiated species do show minor differences up to 2.1%. In contrast, a functionalization with fluorescent labels yields considerably different CCSs, which differ up to 5.5%.
However, as shown for the LeY submotifs LeX vs. BG H2, the above-mentioned trends cannot be generalized and may in some cases even be reversed. Here, the native, underivatized form of the glycan show a difference of 7% in CCS for lithium adducts. Upon modification of the reducing end, the quality of the separation suffers drastically. With up to 3% difference alditol structures may be resolvable on some instruments, while 2-AA, 2-AB and ProA labelled ions cannot be distinguished (<2%). Remarkably, although the trend is reversed for the trisaccharide isomer pairs LeA/BG H1 and LeX vs. BG H2, the average CCS difference is similar at ∼2%. For the tetrasaccharides, the CCS difference is even larger with almost 3%.
To evaluate which CCS difference is sufficient to identify two isomeric species in a mixture, the resolving power from eqn (2) has to be considered. Besides showing the most important factors that influence and, ultimately, determine Rs in DTIMS, eqn (2) also provides a means to calculate the required resolving power (Rp) to achieve a specified peak-to-peak resolution for a given pair of ions. If the relative CCS difference of two ions is 2%, a resolving power of 64 (corresponding to a plate number of 22500) is required to distinguish them (i.e. separation with a peak-to-peak resolution of Rs = 0.75). To achieve baseline resolution for the same two peaks (Rs = 1.5), the resolving power has to be substantially higher, approximately 127 (corresponding to a plate number of 90000). This is already achievable with state-of-the-art custom-built and commercial instruments and shows that IMS can be readily applied for isomer separations, as shown in this study for fucose-containing isomers.
The impact of labelling on fucose migration
The isomer pairs LeA/BG H1 and LeX/BG H2 are known to undergo fucose migration as protonated ions. Fucose migration is a gas-phase rearrangement reaction during which fucose residues relocate within a glycan during a MS experiment. As a result, sequence information can get lost in tandem MS experiments, which can lead to erroneous structural assignments.43 Traditionally, fucose migration was always closely related to a fragmentation via CID.36 However, more recent studies using cold-ion IR spectroscopy showed that fucose migration reactions have a rather low activation barrier and can therefore also occur without dissociation.35,44 In this context, the type of adduct as well as the position of the charge was shown to be crucial.41 Metal adducts generally do not show fucose migration; protonated species on the other hand can rearrange when the proton is mobile and located at a certain position within the glycan.35The results of the isomer pairs LeA/BG H1 and LeX/BG H2 obtained here fully agree with those of previous reports. LeA/BG H1 as well as LeX/BG H2 yield very similar CCSs as protonated ions and the ATDs overlap perfectly, which indicates migration into a similar structure. A similar behaviour can by hypothesized for protonated reduced glycans, which show very similar CCSs that are well within the error of the measurement (relative standard deviation (RSD) of 0.5%).45 However, based on the present data a clear conclusion cannot be drawn. In strong contrast, most of the metal adducts differ substantially in CCS, which clearly contradicts a rearrangement reaction. Labelling with 2-AA, 2-AB and ProA not only changes the UV and fluorescence activity of the glycan, but also introduces apparent basic sites, which reduces or inhibits proton mobility. As a result, different structures leading to distinct CCSs are retained. Fluorescence labelling can therefore not only help to increase isomer separation in IMS as shown above, but can also inhibit fucose migration in protonated glycan ions.
Conclusions
Here we systematically evaluate the impact of reducing end modifications on the CCS and isomer separation of glycans. A set of Lewis and ABO blood group isomers was derivatized using labels established in LC. Their DTCCSsHe were measured in positive and negative ion polarity. Furthermore, the influence of alkali metal salt adduction was evaluated. Our results show that fluorescent labels can significantly influence the gas-phase structure of glycans. As a result, reducing end modifications can considerably improve, but in some cases also diminish the quality of a given isomer separation. Based on the limited set of investigated glycans, no general trends to increase selectivity was observable. Therefore, more glycans have to be analysed in order to predict, which reducing-end modification is required to optimize a particular isomer separation.Seen from a broader perspective, the presented data show the great potential of an LC-IM-MS coupling for glycomics. Both methods have previously shown their individual strengths and weaknesses in glycan analysis. LC can resolve and quantify certain isomers and retention indices (i.e. glucose units46) can be used for the structural identification of known components. IM-MS on the other hand is more sensitive and can also resolve isomers with an amphiphilic character such as synthetic glycans or glycolipids, which due to their mixed polarity can often not be separated by LC.11 In addition, fragmentation and subsequent IMS analysis enables the rapid identification of unknown components based on database CCSs of small fragments.12,13,28 Regarding time scale, LC and IM-MS are furthermore highly complementary and data can be obtained simultaneously on a high-throughput scale.47,48 A combination of LC and IM-MS is therefore highly synergistic and more than the sum of its parts. When combined with suitable software tools to annotate tandem MS spectra and calculate glucose units49 and CCSs,50 LC-IM-MS has the potential to serve as the future core technology in glycomics.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge funding from the German Research Foundation (FOR2177/P02). Open Access funding was provided by the Max Planck Society.References
- A. Varki, Glycobiology, 2017, 27, 3–49 CrossRef CAS PubMed.
- L. Bode, Glycobiology, 2012, 22, 1147–1162 CrossRef CAS PubMed.
- A. Takada, K. Ohmori, T. Yoneda, K. Tsuyuoka, A. Hasegawa, M. Kiso and R. Kannagi, Cancer Res., 1993, 53, 354–361 CAS.
- P. Sozzani, R. Arisio, M. Porpiglia and C. Benedetto, Int. J. Surg. Pathol., 2008, 16, 365–374 CrossRef PubMed.
- J. Hofmann and K. Pagel, Angew. Chem., Int. Ed., 2017, 56, 8342–8349 CrossRef CAS PubMed.
- M. Melmer, T. Stangler, A. Premstaller and W. Lindner, J. Chromatogr. A, 2011, 1218, 118–123 CrossRef CAS PubMed.
- E. Largy, F. Cantais, G. Van Vyncht, A. Beck and A. Delobel, J. Chromatogr., A, 2017, 1498, 128–146 CrossRef CAS PubMed.
- K. A. Morrison and B. H. Clowers, Curr. Opin. Chem. Biol., 2017, 42, 119–129 CrossRef PubMed.
- C. Manz and K. Pagel, Curr. Opin. Chem. Biol., 2018, 42, 16–24 CrossRef CAS PubMed.
- T. Pacini, W. Fu, S. Gudmundsson, A. E. Chiaravalle, S. Brynjolfson, B. O. Palsson, G. Astarita and G. Paglia, Anal. Chem., 2015, 87, 2593–2599 CrossRef CAS PubMed.
- J. Hofmann, H. S. Hahm, P. H. Seeberger and K. Pagel, Nature, 2015, 526, 241–244 CrossRef CAS PubMed.
- H. Hinneburg, J. Hofmann, W. B. Struwe, A. Thader, F. Altmann, D. Varon Silva, P. H. Seeberger, K. Pagel and D. Kolarich, Chem. Commun., 2016, 52, 4381–4384 RSC.
- W. B. Struwe, K. Pagel, J. L. P. Benesch, D. J. Harvey and M. P. Campbell, Glycoconjugate J., 2016, 33, 399–404 CrossRef CAS PubMed.
- M. P. Campbell, R. Peterson, J. Mariethoz, E. Gasteiger, Y. Akune, K. F. Aoki-Kinoshita, F. Lisacek and N. H. Packer, Nucleic Acids Res., 2014, 42, D215–D221 CrossRef CAS PubMed.
- G. Paglia and G. Astarita, Nat. Protoc., 2017, 12, 797–813 CrossRef CAS PubMed.
- K. Pagel and D. J. Harvey, Anal. Chem., 2013, 85, 5138–5145 CrossRef CAS PubMed.
- J. Hofmann, W. B. Struwe, C. A. Scarff, J. H. Scrivens, D. J. Harvey and K. Pagel, Anal. Chem., 2014, 86, 10789–10795 CrossRef CAS PubMed.
- N. M. Lareau, J. C. May and J. A. McLean, Analyst, 2015, 140, 3335–3338 RSC.
- M. Pabst, D. Kolarich, G. Poltl, T. Dalik, G. Lubec, A. Hofinger and F. Altmann, Anal. Biochem., 2009, 384, 263–273 CrossRef CAS PubMed.
- L. Royle, R. A. Dwek and P. M. Rudd, Curr. Protoc. Protein Sci., 2006, 43, 12.6.1–12.6.45 Search PubMed.
- F. Altmann, Anal. Biochem., 1992, 204, 215–219 CrossRef CAS PubMed.
- S. J. Allen, K. Giles, T. Gilbert and M. F. Bush, Analyst, 2016, 141, 884–891 RSC.
- H. E. Revercomb and E. A. Mason, Anal. Chem., 1975, 47, 970–983 CrossRef CAS.
- V. Mantovani, F. Galeotti, F. Maccari and N. Volpi, Electrophoresis, 2016, 37, 1514–1524 CrossRef CAS PubMed.
- A. Varki, R. D. Cummings, M. Aebi, N. H. Packer, P. H. Seeberger, J. D. Esko, P. Stanley, G. Hart, A. Darvill, T. Kinoshita, J. J. Prestegard, R. L. Schnaar, H. H. Freeze, J. D. Marth, C. R. Bertozzi, M. E. Etzler, M. Frank, J. F. Vliegenthart, T. Lutteke, S. Perez, E. Bolton, P. Rudd, J. Paulson, M. Kanehisa, P. Toukach, K. F. Aoki-Kinoshita, A. Dell, H. Narimatsu, W. York, N. Taniguchi and S. Kornfeld, Glycobiology, 2015, 25, 1323–1324 CrossRef CAS PubMed.
- R. B. Myers, S. Srivastava and W. E. Grizzle, J. Urol., 1995, 153, 1572–1574 CrossRef CAS PubMed.
- J. Pendu, S. Marionneau, A. Cailleau-Thomas, J. Rocher, B. Moullac-Vaidye and M. Clément, APMIS, 2001, 109, 9–26 CrossRef PubMed.
- J. Hofmann, A. Stuckmann, M. Crispin, D. J. Harvey, K. Pagel and W. B. Struwe, Anal. Chem., 2017, 89, 2318–2325 CrossRef CAS PubMed.
- L. Veillon, Y. Huang, W. Peng, X. Dong, B. G. Cho and Y. Mechref, Electrophoresis, 2017, 38, 2100–2114 CrossRef CAS PubMed.
- D. Reusch, M. Haberger, B. Maier, M. Maier, R. Kloseck, B. Zimmermann, M. Hook, Z. Szabo, S. Tep, J. Wegstein, N. Alt, P. Bulau and M. Wuhrer, mAbs, 2015, 7, 167–179 CrossRef CAS PubMed.
- T. Keser, T. Pavic, G. Lauc and O. Gornik, Front. Chem., 2018, 6, 324 CrossRef PubMed.
- D. Reusch, M. Haberger, D. Falck, B. Peter, B. Maier, J. Gassner, M. Hook, K. Wagner, L. Bonnington, P. Bulau and M. Wuhrer, mAbs, 2015, 7, 732–742 CrossRef CAS PubMed.
- E. Lattova, S. Snovida, H. Perreault and O. Krokhin, J. Am. Soc. Mass Spectrom., 2005, 16, 683–696 CrossRef CAS PubMed.
- S. Zhou, L. Veillon, X. Dong, Y. Huang and Y. Mechref, Analyst, 2017, 142, 4446–4455 RSC.
- M. Lettow, E. Mucha, C. Manz, D. A. Thomas, M. Marianski, G. Meijer, G. von Helden and K. Pagel, Anal. Bioanal. Chem., 2019, 411, 4637–4645 CrossRef CAS PubMed.
- M. Wuhrer, C. A. Koeleman, C. H. Hokke and A. M. Deelder, Rapid Commun. Mass Spectrom., 2006, 20, 1747–1754 CrossRef CAS PubMed.
- D. J. Harvey, J. Am. Soc. Mass Spectrom., 2000, 11, 900–915 CrossRef CAS PubMed.
- D. J. Harvey, Mass Spectrom. Rev., 2017, 36, 255–422 CrossRef CAS PubMed.
- L. R. Ruhaak, G. Zauner, C. Huhn, C. Bruggink, A. M. Deelder and M. Wuhrer, Anal. Bioanal. Chem., 2010, 397, 3457–3481 CrossRef CAS PubMed.
- Y. Huang and E. D. Dodds, Anal. Chem., 2013, 85, 9728–9735 CrossRef CAS PubMed.
- W. B. Struwe, C. Baldauf, J. Hofmann, P. M. Rudd and K. Pagel, Chem. Commun., 2016, 52, 12353–12356 RSC.
- W. B. Struwe, J. L. Benesch, D. J. Harvey and K. Pagel, Analyst, 2015, 140, 6799–6803 RSC.
- R. W. Vachet, B. M. Bishop, B. W. Erickson and G. L. Glish, J. Am. Chem. Soc., 1997, 119, 5481–5488 CrossRef CAS.
- E. Mucha, M. Lettow, M. Marianski, D. A. Thomas, W. B. Struwe, D. J. Harvey, G. Meijer, P. H. Seeberger, G. von Helden and K. Pagel, Angew. Chem., Int. Ed., 2018, 57, 7440–7443 CrossRef CAS PubMed.
- S. M. Stow, T. J. Causon, X. Zheng, R. T. Kurulugama, T. Mairinger, J. C. May, E. E. Rennie, E. S. Baker, R. D. Smith, J. A. McLean, S. Hann and J. C. Fjeldsted, Anal. Chem., 2017, 89, 9048–9055 CrossRef CAS PubMed.
- L. Royle, C. M. Radcliffe, R. A. Dwek and P. M. Rudd, in Glycobiology Protocols, ed. I. Brockhausen, Humana Press, Totowa, NJ, 2007, pp. 125–143, DOI:10.1385/1-59745-167-3:125.
- L. Royle, M. P. Campbell, C. M. Radcliffe, D. M. White, D. J. Harvey, J. L. Abrahams, Y. G. Kim, G. W. Henry, N. A. Shadick, M. E. Weinblatt, D. M. Lee, P. M. Rudd and R. A. Dwek, Anal. Biochem., 2008, 376, 1–12 CrossRef CAS PubMed.
- A. Shubhakar, K. R. Reiding, R. A. Gardner, D. I. Spencer, D. L. Fernandes and M. Wuhrer, Chromatographia, 2015, 78, 321–333 CrossRef CAS PubMed.
- C. M. Radcliffe, L. Royle, M. P. Campbell, P. M. Rudd and R. A. Dwek, Bioinformatics, 2008, 24, 1214–1216 CrossRef PubMed.
- M. T. Marty, A. J. Baldwin, E. G. Marklund, G. K. Hochberg, J. L. Benesch and C. V. Robinson, Anal. Chem., 2015, 87, 4370–4376 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9an00937j |
| This journal is © The Royal Society of Chemistry 2019 |