
Semi-automatic instrumentation for nucleic acid extraction and purification to quantify pathogens on surfaces†
 *a
and
Junhong
Min
*a
*a
and
Junhong
Min
*a
Abstract
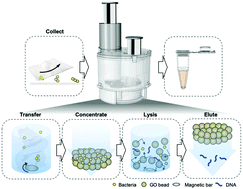
Public lavatories may cause the spread of infectious pathogens because they are enclosed spaces that both healthy people and patients can use. Thus, surface analysis for microbial contamination in public lavatories is of great importance because it is considered as an indicator of hygiene control. Herein, we developed polymerase chain reaction (PCR)-compatible surface sample preparation tools to increase the detection sensitivity and reproducibility within a short time using a semi-automatic detection system. The bacteria and viruses on different surfaces were collected using half A4-sized wipes. The wipes were treated through four different processes in a cartridge: (1) the pathogens were transferred from the wipes to the aqua phase using simple gentle vortexing; (2) the bacteria and viruses were concentrated by adsorption on the graphene surface; (3) the pathogens on the graphene layer were perfectly lysed using bead-beating tools and (4) the released DNA/RNA was collected in a microtube. The prepared nucleic acid sample was amplified using PCR or loop-mediated isothermal amplification (LAMP). At least one order of magnitude higher sensitivity was achieved using the wipe collecting method compared to that achieved using the normal swab method. This was confirmed using a semi-automatic cartridge for the wipe sampling in a lavatory hygiene test.
- This article is part of the themed collections: Coronavirus articles - free to access collection and Coronavirus collection – Analytical
 Please wait while we load your content...
Please wait while we load your content...

