
Surface-enhanced Raman spectroscopy of microorganisms: limitations and applicability on the single-cell level†
Ruben
Weiss
 a,
Márton
Palatinszky
b,
Michael
Wagner
b,
Reinhard
Niessner
a,
Martin
Elsner
a,
Michael
Seidel
a and
Natalia P.
Ivleva
*a
a,
Márton
Palatinszky
b,
Michael
Wagner
b,
Reinhard
Niessner
a,
Martin
Elsner
a,
Michael
Seidel
a and
Natalia P.
Ivleva
*a
aTechnical University of Munich, Institute of Hydrochemistry, Chair of Analytical Chemistry and Water Chemistry, Marchioninistrasse 17, D-81377 Munich, Germany. E-mail: natalia.ivleva@ch.tum.de; Fax: +49 89 2180 78255; Tel: +49 89 2180 78252
bUniversity of Vienna, Department of Microbiology and Ecosystem Science, Division of Microbial Ecology, Research Network “Chemistry meets Microbiology”, Althanstrasse 14, A-1090 Vienna, Austria
First published on 17th December 2018
Abstract
Detection and characterization of microorganisms is essential for both clinical diagnostics and environmental studies. An emerging technique to analyse microbes at single-cell resolution is surface-enhanced Raman spectroscopy (surface-enhanced Raman scattering: SERS). Optimised SERS procedures enable fast analytical read-outs with specific molecular information, providing insight into the chemical composition of microbiological samples. Knowledge about the origin of microbial SERS signals and parameter(s) affecting their occurrence, intensity and/or reproducibility is crucial for reliable SERS-based analyses. In this work, we explore the feasibility and limitations of the SERS approach for characterizing microbial cells and investigate the applicability of SERS for single-cell sorting as well as for three-dimensional visualization of microbial communities. Analyses of six different microbial species (an archaeon, two Gram-positive bacteria, three Gram-negative bacteria) showed that for several of these organisms distinct features in their SERS spectra were lacking. As additional confounding factor, the physiological conditions of the cells (as influenced by e.g., storage conditions or deuterium-labelling) were systematically addressed, for which we conclude that the respective SERS signal at the single-cell level is strongly influenced by the metabolic activity of the analysed cells. While this finding complicates the interpretation of SERS data, it may on the other hand enable probing of the metabolic state of individual cells within microbial populations of interest.
Introduction
In the last decade surface-enhanced Raman scattering (SERS) has received increasing attention for the analysis of biological samples, ranging from environmental studies1–4 to diagnostics.5–8 The underlying principle of Raman spectroscopy (RS) is based on the inelastic scattering of light giving rise to characteristic spectra that yield chemical information in a non-invasive, non-destructive way with little interference of water. Combining the band-related data of the spectrometer with the spatial information of a confocal microscope, enables analyses of sample material with a spatial resolution in the micrometre-range necessary for studying the chemical composition of microbes on a single-cell level. The resolution is physically limited by the diffraction defined through the excitation wavelength and numerical aperture of the microscope objective. Thus, with RS the analysis of single microbial cells under natural conditions is possible. If combined with stable isotope probing, the activity of microbial cells within their habitat can be deduced from red-shifts of certain peaks in the spectra of active cells9,10 and if desired such analyses can be combined with identification of the respective organisms with fluorescence in situ hybridization (FISH).11 Excitingly, recent technological developments even allow sorting of individual microbial cells with selected characteristics within their Raman spectra for subsequent genomic analysis or cultivation.12Sorting of single cells by means of optical trapping and discrimination by RS (laser tweezer Raman spectroscopy: LTRS or Raman tweezers) was introduced by Xie, Chen and Li13 and applied in several studies. Huang et al. showed that single cells of unlabelled Escherichia coli and 13C-labelled Pseudomonas fluorescens from a mixed population can be isolated by Raman tweezers.14 A recent study by Berry et al. used LTRS to detect incorporation of deuterium into cecal microbiota of mice as a marker for metabolic activity.9 Subsequently, genomic information of sorted microbial cells was analysed by whole genome amplification and sequencing. Wang et al. introduced another method for the sorting of single cells by RS in combination with laser-induced forward transfer and termed the technique Raman activated cell ejection (RACE).15
In conventional RS, the analytical throughput is limited because of the low scattering cross section. SERS offers an improvement with enhancement factors of up to 1011.16 The underlying principle is that surfaces of nanosized metal structures (Ag or Au) can enhance the local electric field of the excitation source because of surface plasmon resonance. Scattering intensities increase since the Raman effect depends on the electric field strength. Also, the polarizability of molecules directly attached to these metal surfaces changes, enhancing the resultant Raman scattering.
In 1998 Efrima and Bronk showed the general applicability of SERS for the analysis of bacteria.17 Species- and even strain-specific characterization was demonstrated by Jarvis and Goodacre in 2004.18 In 2007 Kahraman et al. illustrated that the SERS analysis down to a few bacterial cells can be realised.19 Kubryk et al. presented 2015 a reproducible SERS analysis of bacteria at the single-cell level with the capability of detecting stable isotope incorporation.20 The characteristics of microbial SERS spectra were mainly attributed to flavins21 and peptidoglycan.18 Recently, major contributions of adenine-derived compounds were recognised with a marker band at about 730 cm−1.22,23 Controversially, Chisanga et al. applying a different sample preparation protocol observed the absence of this prominent SERS peak.24 Premasiri et al. reported that SERS spectra of bacteria display the salvaging pathways and secretion of purines.23 Molecules, which contribute to the SERS signal and only occur intracellular or are anchored to the cell envelope, are not complicating the analyses. In contrast, extracellular released compounds detected by SERS render the assignment of a SERS signal to defined cells very challenging. Degradation pathways, secretion/excretion processes and shock reactions heavily depend on the physiological state of the microorganisms and might also be influenced by the sample preparation. Therefore, studies are needed to clarify the mechanism causing SERS signals of microbial samples and to address the factors that influence the composition of resulting spectra which might – in the worst case – lead to erroneous results. In 2015 Zhou et al. reported that the differentiation of live and dead bacteria is possible by SERS because of disappearing signals for dead cells.25 In the present study, we confirm this observation but critically evaluate whether the missing peaks may be used as an indicator for dead bacteria or rather as evidence for metabolic inactivity.
In the first part of this work, SERS signals from six phylogenetically diverse microorganisms representing different cell envelope types including an archaeon and Gram-positive as well as Gram-negative bacteria were analysed to investigate the variability and reproducibility of SERS for the analyses of microbes. In addition, to outline potential limitations, the Gram-negative model bacterium Escherichia coli was chosen to study the effect of varying physiological conditions such as storage times and growth in heavy water on the SERS signal. Finally, in the second section of this study, the applicability of SERS for LTRS of microbial cells and for three-dimensional analyses of biological samples was evaluated.
Experimental
Chemicals and materials
Standard chemicals were purchased from Carl Roth GmbH & Co. KG (Karlsruhe, Germany), Merck KGaA (Darmstadt, Germany) or Sigma-Aldrich (Vienna, Austria; Steinheim, Germany). 13C6-D-glucose (13C6H12O6) and D2O were obtained from Sigma-Aldrich. The Colilert-18 test kit was purchased from IDEXX Europe B.V. (Hoofddorp, Netherlands) and the LIVE/DEAD BacLight kit from Life Technologies GmbH (Darmstadt, Germany). Only purified water was used (Milli-Q, Merck KGaA). Glass capillaries with internal dimensions of 100 μm × 1 mm × 50 mm and a wall thickness of 70 μm were purchased from CM Scientific (Silsden, United Kingdom).Microorganisms
Bacterial strains were either obtained from the German Collection of Microorganisms and Cell Cultures (DSMZ, Braunschweig, Germany) or LGC Standards GmbH (Wesel, Germany). Bacillus subtilis (DSM 1087), Escherichia coli (DSM 498; DSM 1116), Micrococcus luteus (DSM 20030) and Pseudomonas putida (ATCC 12633) were cultured aerobically with LB and a supplemented M9 minimal medium as biological triplicates to the stationary phase (see ESI Fig. S1a†). Nitrospira inopinata (DSM 105286)26 and Nitrososphaera gargensis (DSM 103042)27,28 were obtained from culture collection of the Division of Microbial Ecology at the University Vienna but are also available from the DSMZ. These strains were cultured in a mineral salt medium with ammonium as energy source as described previously.28 Detailed procedures on the cultivation are included in the ESI.†In situ sample preparation for SERS analysis
The silver colloid synthesis applied in this study is based on the method by Leopold and Lendl.29 It was performed in situ30 and followed the modified procedure as described elsewhere.20 Microbial cultures were either used directly or stored at 5 °C up to 5 days before silver nanoparticle (AgNP) synthesis. As this synthesis was performed equally 1 h prior to SERS measurements we disregarded the reported disadvantageous behaviour of toxic silver31 for the SERS analysis of bacteria. It is irrelevant for the findings of our study if the analysed microorganisms are killed during the in situ AgNP synthesis. Surprisingly, cultivation experiments with treated E. coli cells show still microbial growth (comp. ESI Fig. S1a†). Although E. coli strain W is non-mucoid it is known to form capsular polysaccharides at temperatures below 15 °C.32 Therefore, microbial cells were washed before nanoparticle synthesis to avoid interferences with the colloid synthesis. For this purpose, 1 ml of the culture was centrifuged (5500g, 1 min, 20 °C), the supernatant discarded and the cell pellet resuspended with water. This washing step was repeated twice. After the third centrifugation, the cell pellet was resuspended with 450 μl of a reducing solution. For SERS analysis with a 633 nm laser the reducing solution was prepared by dissolving 11.6 mg NH2OH·HCl in 96.7 ml water and adding 3.3 ml of a 0.1 M sodium hydroxide solution. Finally, 50 μl a 10 mM silver nitrate solution was added to the resuspended microbial cells at room temperature to form AgNP on the cell surface. For SERS analysis with an excitation wavelength of 532 nm the concentration of all reagents was doubled and colloidal in situ synthesis was performed at 5 °C. The reagents were mixed by inverting the sample three times. The sample colour changed instantly to yellowish-grey indicating the formation of silver nanoparticles (comp. ESI Fig. S2†). For dried samples, 1 μl of the cell suspension was spotted onto a glass slide after a reaction time of 1 h if not stated otherwise. The spot was dried at room temperature for at least 30 min. For the artificial biofilm, a 2% (m/m) suspension of agarose was stirred under reflux at 90 °C for 5 min. Subsequently, at 37 °C 300 μl of each 12C- and 13C-labelled colloidal sample were added to 8.4 ml of the dissolved agarose, gently pivoted and casted (2 ml) into a preheated Petri dish with a diameter of 5 cm. The resulting gels were covered with water. For the tweezing experiments with a mixture of 12C/13C-labelled cells 0.5 μl of each separately prepared sample was used. In accordance to Berry et al.9 a small volume of this suspension was drawn into rectangular glass capillaries which was already filled to about three quarters with a phosphate-buffered saline (PBS) with 5.6 mg L−1 Tween 20. Particles could be moved through this interface between the two compartments to the cell-free region of the PBS buffer with the activated tweezer laser by moving the capillary with the motorised microscope stage. The inlets of the capillary were sealed with petrolatum.Raman microscopic analysis
All SERS analyses were performed with an LabRAM HR800 microspectrometer (Horiba Jobin Yvon, France) equipped with a frequency-doubled Nd:YAG laser (532 nm, 35 mW at the sample), a He–Ne laser (633 nm, 13.8 mW at the sample), a standard Nd:YAG laser for optical trapping (1064 nm, 500 mW at the sample), corresponding edge filters, a confocal pinhole set to a diameter of 100 μm, diffraction gratings with 300 or 600 lines per mm and a motorized microscope stage. The stage could be moved while optically trapping cells to sort them. The system was calibrated onto the first-order phonon band of silicon at 520.7 cm−1. Dried samples were measured with a 100× objective (Olympus MPlan N, NA = 0.9). The laser beam was focused onto single cells with an acquisition time of 3 × 1 s and an intensity of 0.1% or 1% of the full laser power. A typical microscopic image of this kind of sample without and with laser illumination (see ESI Fig. S3a/b†) shows experimental evidence for the analysis at the single-cell level by SERS. The SERS spectra of different microorganisms presented in Fig. 1 and the spectra obtained from the optical tweezing setup were measured with the 532 nm laser and the grating with 300 lines per mm in one measurement window resulting in a spectral range of 400–3300 cm−1. The optical trapping measurements were performed with a 63× water immersion objective (Zeiss C-APOCHROMAT, NA = 1.2), 100 ms acquisition time, full power of the tweezing laser (500 mW) and reduced power of the excitation laser (3 mW). All other SERS spectra were acquired with an excitation wavelength of 633 nm and the grating with 600 lines per mm. The artificial biofilm was measured with a 60× water immersion objective (Olympus LUMPlanFL N, NA = 1.0), measurement time of 2 × 100 ms, laser intensity of 1.38 mW and static spectral region of 50–1260 cm−1 and a spatial measurement interval of 1 μm. All spectra related to the storage series of E. coli (DSM 1116) were measured under equal conditions with the 633 nm laser, an effective laser power of 138 μW and five consecutive measurement windows resulting in the spectral range of 50–4000 cm−1. In this measurement series 10 spectra of randomly chosen cells were acquired for each group of biological triplicates, respectively 30 spectra for the biological triplicates cultured with LB medium and a storage time of zero or four days. The evaluation of the non-normalised data from this series was performed automatically by MATLAB R2016a (The MathWorks, USA) in the spectral region of 700–760 cm−1 by fitting a Lorentzian with a linear background. The signal intensity of the SERS peak at 733 cm−1 was determined as the amplitude of the Lorentzian and is in all graphs simply referred to as SERS signal. The SERS spectra are marked as positive hits if the amplitude is bigger than three times the noise averaged over all spectra of one data point plus the baseline at the fitted peak (see ESI Fig. S3c†). The noise was calculated in the Raman silent region at 2500–2600 cm−1. Spectra in Fig. 1 were baseline-corrected with a linear assumption and normalised to maximal intensity between 2900–2960 cm−1 with LabSpec 6 (Horiba Jobin Yvon, France).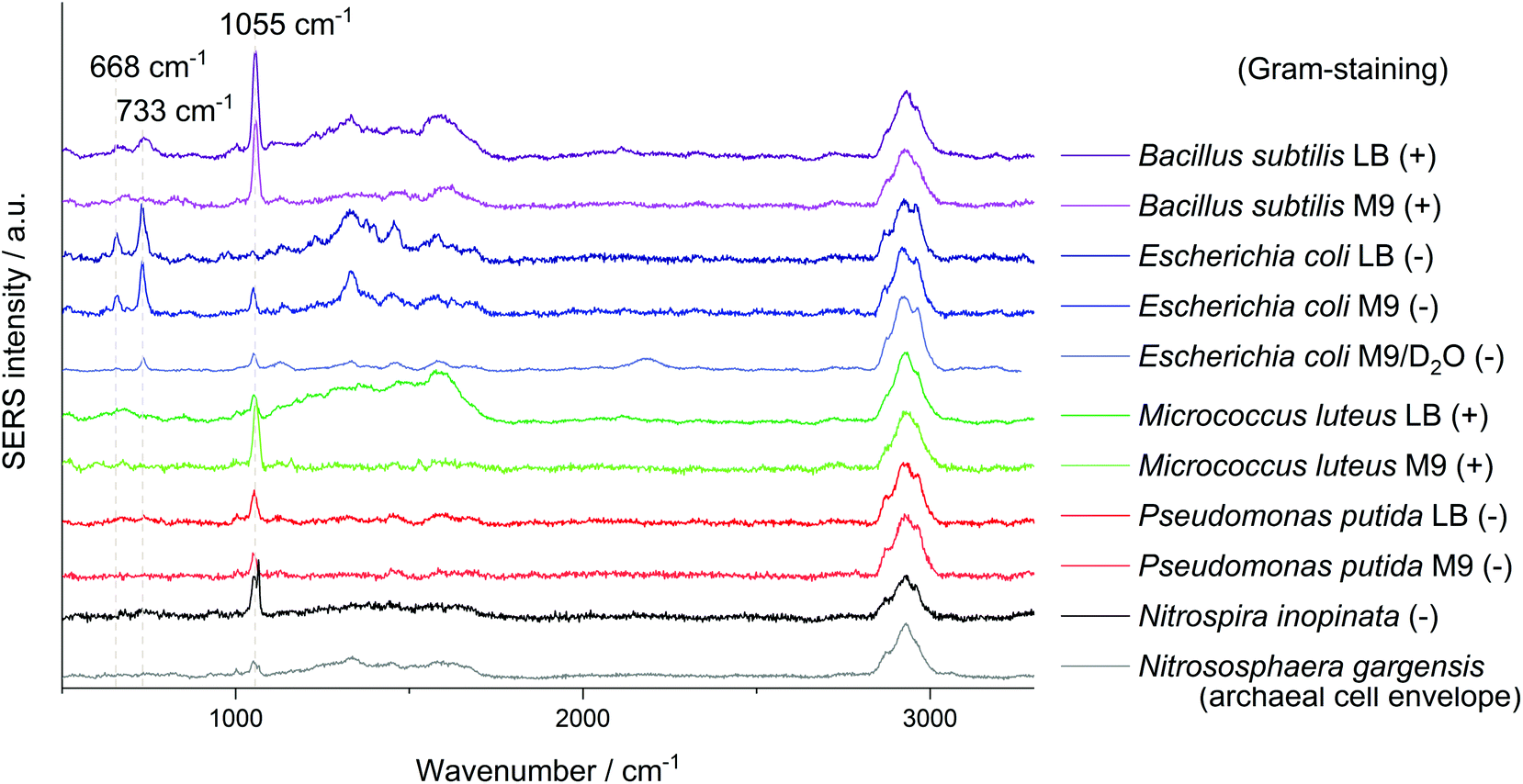 | ||
| Fig. 1 SERS mean spectra of at least 8 single cells of B. subtilis, E. coli, M. luteus, P. putida, N. gargensis and N. inopinata measured with an excitation wavelength of 532 nm. Spectra are baseline-corrected and normalised to the C–H stretching vibration, (+) and (−) indicate Gram-positive and -negative, respectively. M9 and LB refer to the different media used for cultivation of the respective species. D2O indicates that the organism was cultured with heavy water in the medium. The signal at 1084 cm−1 for the complete nitrifier N. inopinata most probably results from precipitated CaCO3 of the medium. | ||
Scanning electron microscopy
Samples were prepared in the same way as for the SERS preparation. The suspension was spotted directly onto the aluminium sample holder and dried at room temperature. A Sigma 300 VP scanning electron microscope (Carl Zeiss AG, Germany) was used. Images were recorded with the In-Lens detector, an acceleration voltage of up to 10 kV and a working distance between 2.8–3.1 mm.Results and discussion
Limitations of SERS for microbial detection
Another signal shared by the spectra of several microorganisms lies at about 1055 cm−1 which was so far mostly attributed to amine but also to phosphate (FAD),24 carbohydrate37 and nitrate38 interacting with AgNP. Simple mixing experiments of AgNO3 with NaNO3 indicate that the Raman peaks of these nitrates at 1044 cm−1 and 1067 cm−1 combine to a broader band in between. The relatively high sodium concentration from the added sodium hydroxide for in situ nanoparticle synthesis might explain the position of the nitrate peak in SERS spectra with AgNO3 as precursor. The reference spectra of other nitrates such as KNO3 or Ca(NO3)2 show an intense signal at exactly 1050 cm−1. As this band mainly occurs when AgNP were synthesised with hydroxylamine as reducing agent it might be related to an excess of hydroxylamine binding to the AgNP. Nitrate from the precursor AgNO3 thermodynamically tends to comproportionate with hydroxylamine, diminishing the band at 1055 cm−1 over time. As the AgNP synthesis is performed in a complex sample, trace elements might catalyse the decomposition of the reducing agent. Still it is surprising that for E. coli, especially if cultured in LB medium, the presumably unspecific peak at 1055 cm−1 is significantly weaker. This could be related to the appearance of new peaks and a competitive binding of analytes with a higher affinity for AgNP.
The intense SERS signals observed for E. coli at 733 cm−1 and 668 cm−1 show the greatest variation in intensity between the investigated microorganisms (Fig. 1). In 2016 Kubryk et al.22 and Premasiri et al.23 assigned these bands to purine bases and biochemically relevant derivatives, e.g., adenine, guanine, hypoxanthine, xanthine, where Premasiri et al. found a perfect alignment of bacterial SERS spectra and these compounds. Earlier the frequently arising SERS peak at 733 cm−1 had also been attributed to N-acetyl-D-glucosamine,18 methylene rocking vibrations,39 a glycosidic ring18 and pyrophosphate mode40 and was also thought to be loosely related to amino acids and phospholipids.41 Surprisingly, a recent study by Chisanga et al. did not observe this signal for E. coli.24 Differences to our methods include the reducing agent (borohydride instead of hydroxylamine) as well as storing of the prepared bacterial cells at −80 °C prior to in situ AgNP synthesis. We observed the signal at 733 cm−1 both with an excitation wavelength of 532 nm (3 s, 35 μW) and 633 nm (3 s, 138 μW) with similar spectral characteristics as presented by Premasiri et al. who used Au-nanoparticle-covered substrates and a 785 nm laser but log-phase cultures.23 However, the signals at 733 cm−1 and 668 cm−1 were not observed for the other five microorganisms analysed in our work at the given measurement conditions. Premasiri et al. pointed out that the interaction time of metallic nanostructure and bacterial cells can play an important role for the occurrence of bacterial SERS signals and related this to the delayed enzymatic degradation and following secretion of metabolites (purines).23 In our case intense SERS signals at 733 cm−1 and 668 cm−1 were observed for E. coli only, irrespective of the reaction time (up to 2 h). Other tested microbes were lacking this characteristic spectral information. Either these organisms do not release the molecules causing SERS signals at 733 cm−1 and 668 cm−1 to the same extent or other properties intervene with the applied preparation protocol. High variations between the single-cell spectra, which are included in Fig. S5/S6 of the ESI,† additionally limit the applicability of SERS to distinguish the microorganisms. Hence, the bands at 733 cm−1 and 668 cm−1 do not seem to be suited for all microorganisms, at least not with the excitation wavelength of 532 nm.
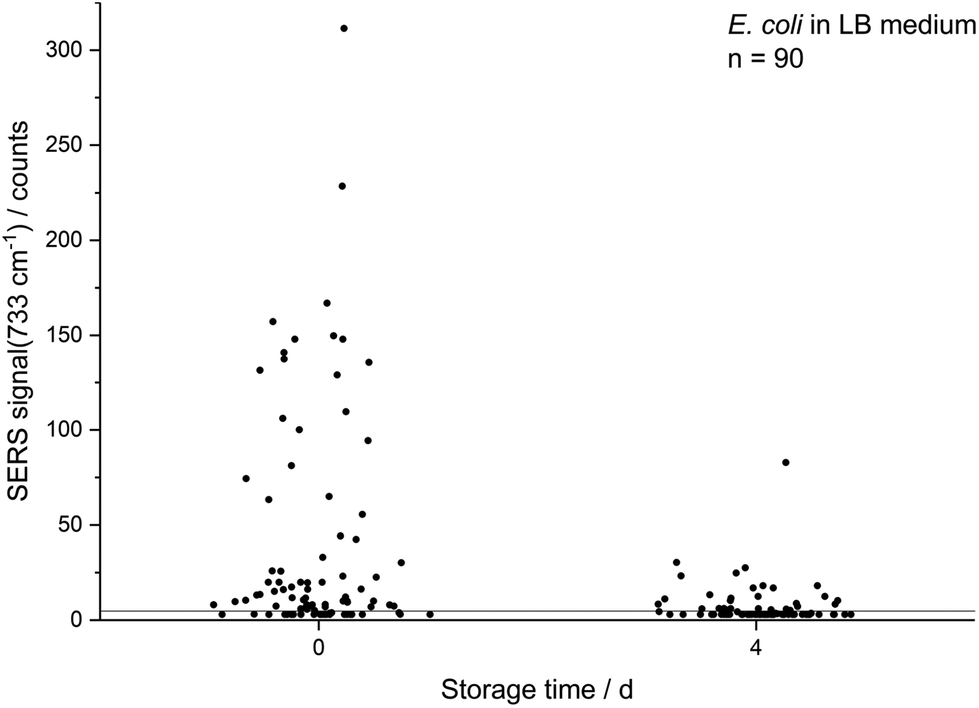 | ||
| Fig. 2 Decreasing absolute SERS intensity at 733 cm−1 of single E. coli (LB medium) cells which were cultured to the stationary phase and either directly subjected to in situ AgNP synthesis or first stored for 4 d. Values are scattered on the abscissa for better readability. | ||
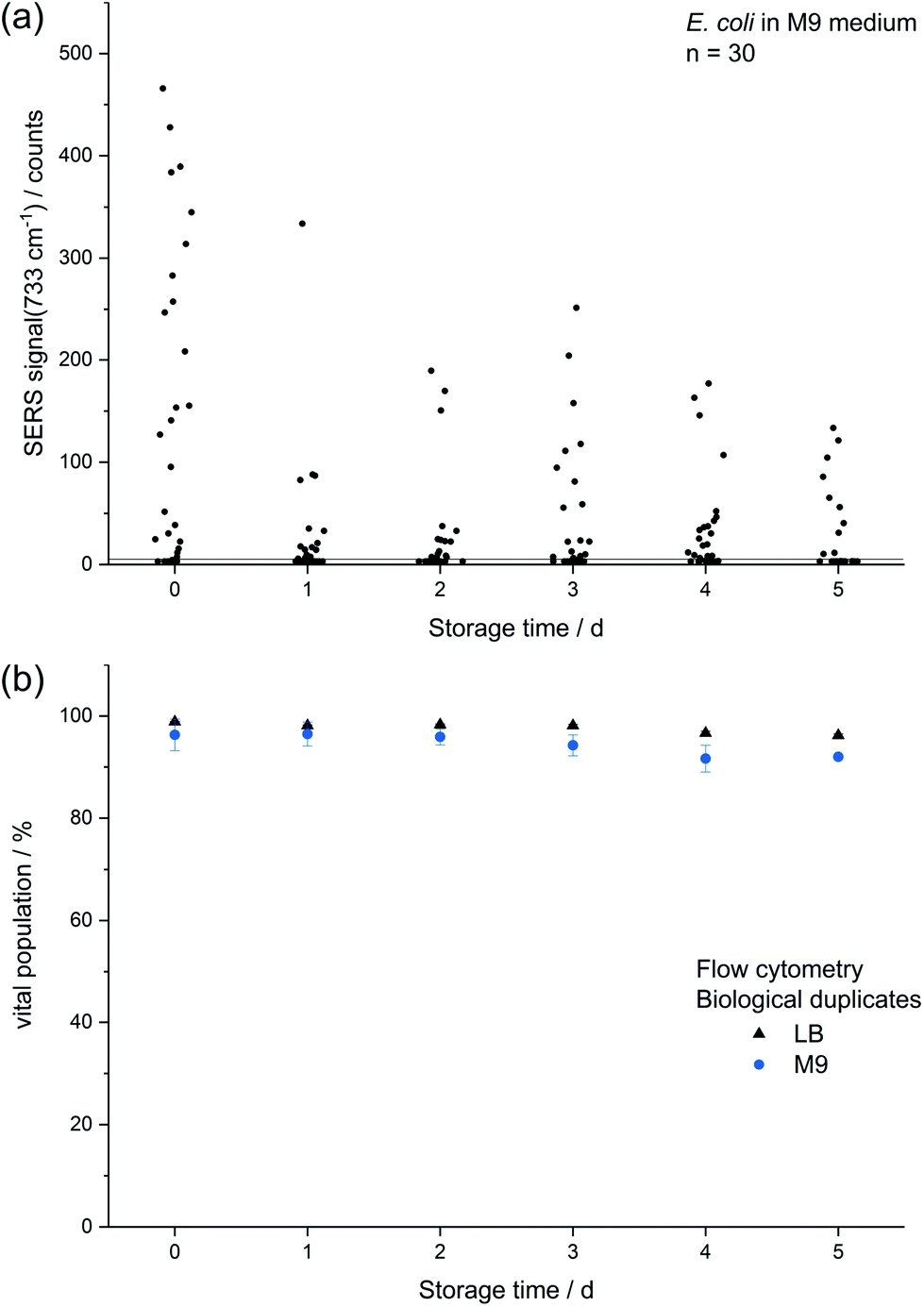 | ||
| Fig. 3 Comparison of (a) absolute SERS signal at 733 cm−1 of single E. coli cells which were cultured to the stationary phase and either directly subjected to in situ AgNP synthesis or first stored up to 5 d and (b) viability of E. coli measured by flow cytometry with two DNA intercalating dyes (propidium iodide: dead, SYTO 9: viable) after 0–5 d storage. | ||
| Storage time/d | Percentage of SERS hits ± 1 s/% | Percentage of SERS hits ± 1 s/% |
|---|---|---|
| LB medium | M9 medium | |
| 0 | 72.2 ± 8.4 | 80.0 ± 10.0 |
| 1 | 46.7 ± 11.5 | 46.7 ± 23.1 |
| 2 | 30.0 ± 34.6 | 50.0 ± 10.0 |
| 3 | 40.0 ± 26.5 | 56.7 ± 35.1 |
| 4 | 50.0 ± 17.3 | 63.3 ± 15.3 |
| 5 | 32.2 ± 19.5 | 33.3 ± 20.8 |
The decreasing SERS signal may be caused by two reasons: (1) the AgNP are not in close vicinity to microbial cells after sample preparation is finished. (2) The analyte(s) itself is(are) not present in the same condition or concentration. The first assumption was already considered for dead bacteria by Zhou et al. who reported that the signal at 735 cm−1 decreased if E. coli cells were exposed to antibiotics as well as if varying percentages of autoclaved cells were added and explained this finding by a vanishing surface charge.25 Indeed, also in the current study we did not observe any SERS signals for heat-killed E. coli cells when applying the same measurement parameters (data not shown). To test the hypothesis that the decrease in SERS signal could be attributable to the death of cells during storage we analysed the viability of E. coli by flow cytometry using the dyes propidium iodide (emission at 617 nm) and SYTO 9 (emission at 501 nm). These stains predominantly intercalate with nucleic acids of dead and living cells, respectively. Fig. 3b shows that the viability of the stored E. coli decreased only marginally by less than 5%. Therefore, flow cytometric analysis does not confirm the hypothesis that the decline in SERS signal is caused by dead bacteria. This result is in agreement with Colilert tests which revealed no noticeable drop of viability of the E. coli cells induced by the applied storage. We found more than 109 CFU ml−1 in all the samples cultured in LB medium and 2 × 108 CFU ml−1 in all of those in M9 medium (see Fig. S1b†).
Scanning electron microscopy (SEM) measurements were performed to investigate the hypothesis that missing AgNP were the reason for decreasing SERS signals. Fig. 4 illustrates that there is no significant difference for microbial samples which were either directly subjected to the in situ AgNP synthesis (a) or first stored for 12 d (b). Thus, we conclude that it must be the target analyte detected by SERS which disappears over storage time or can no longer be released by stored E. coli cells leading to the declining SERS signals. This finding was validated by mass spectrometric analysis of the metabolite adenine in the supernatant of washed E. coli cells. As shown in Fig. S7† the signal of adenine declines for samples of stored cultures in comparison to freshly harvested, washed cells. Further, Chiu et al. recently report a rapid but over time decreasing release of purines by bacteria upon transferring them from a nutrient-rich into a nutrient-deprived environment.42
 | ||
| Fig. 4 SEM images of E. coli cultured with M9 medium after 0 d (a) and 12 d (b) storage and following AgNP in situ synthesis. | ||
In bacteria approaching the stationary phase purine consumption by macromolecular synthesis abruptly slows.43 Additionally, the cells start to break down free messenger ribonucleic acid (mRNA), transfer RNA (tRNA) and especially ribosomal RNA (rRNA).44 This leads to a cellular accumulation of the end products of this degradation process including purine derivatives. The chemical nature of these products depends on the purine metabolic pathways present in the respective species, which vary considerably between bacterial species.23 For E. coli the main end products will be adenine, xanthine, and hypoxanthine which are partially excreted. This accumulation and excretion happen within a time frame of about an hour after transferring the cells from medium into water or glucose deprived medium and is facilitated by the fact that hypoxanthine phosphoribosyl transferases operate close to the membrane and also transport their products through the membrane. The secretion of the purine derivatives might act as a signal for the culture to go into dormancy.44 In addition to these phenomena, cyclic adenosine monophosphate is upregulated and also secreted in E. coli during glucose starvation and will thus contribute to the pool of extracellular purines in starved cultures. Furthermore, Bayer reported reversible cell wall disruptions upon hypoosmotic shock and the associated release of intracellular substances, e.g. acid-soluble nucleotides.45 This starvation- and/or shock-induced breakdown and release of low-molecular weight nucleotides or degradation products well explains the characteristic E. coli SERS signal at 733 cm−1. The phenomenon is able to relate our observations to a metabolic state instead of pure surface properties. Consequently, cells of microbial species like E. coli responding with these mechanisms to starvation or physiological stress (as induced by the cell washing procedure before nanoparticle synthesis) would be detectable by SERS if they are metabolically active to this point. Conversely, metabolically inactive and dormant cells would not release purine derivatives. Hence, they would show no or weak SERS signals even though they are viable, explaining the results obtained with stored E. coli cells. Furthermore, it is well known that several slow growing microorganisms (including nitrifiers such as N. inopinata and N. gargensis) show a much slower decrease in rRNA in response to starvation or stress46–49 and might thus not exhibit the characteristic SERS signal at 733 cm−1 even if they were metabolically active at the start of the cell washing and nanoparticle formation procedure.
SERS of deuterated microbial cells
In addition to storage, also cultivation conditions seem to influence the results of the E. coli SERS analysis. Fig. 5a shows a comparison of E. coli cultured with M9 medium containing only normal water and with 25% (v/v) heavy water for 16 h, respectively. The red-shift up to 720 cm−1 with a mean at 728 cm−1 can be explained by the partial substitution of 1H with D in the purine bases and their derivatives. The SERS intensity of the fitted peak at about 733 cm−1 is significantly reduced for the deuterated cells. This effect is not expected to be related to a direct major impact onto the growth of E. coli as the growth curve of E. coli grown in media produced with concentrations of up to 25% of D2O does not change fundamentally.9,50 Surprisingly the position of the SERS purine bases band for deuterated cells varies stronger in comparison to unlabelled cells. The higher variance results either from varying amounts of incorporated deuterium into the purine bases, from changing positions of incorporated deuterium in purine bases or from a fundamental impact of heavy water onto the SERS causing mechanism, e.g., the composition of released compounds. In any way, this indicates that conclusions onto the percentage of incorporated deuterium by certain microbial species based on this SERS peak is not as straight forward as for the stable isotopes 14N/15N.4 The change of the SERS signal intensity of deuterated cells caused by storage over 5 d (Fig. 5b) is negligible in contrast to experiments with unlabelled E. coli cells (Fig. 3a). Thus, we suggest that either the formation or the release of the SERS target compound(s) is reduced because of incorporated deuterium.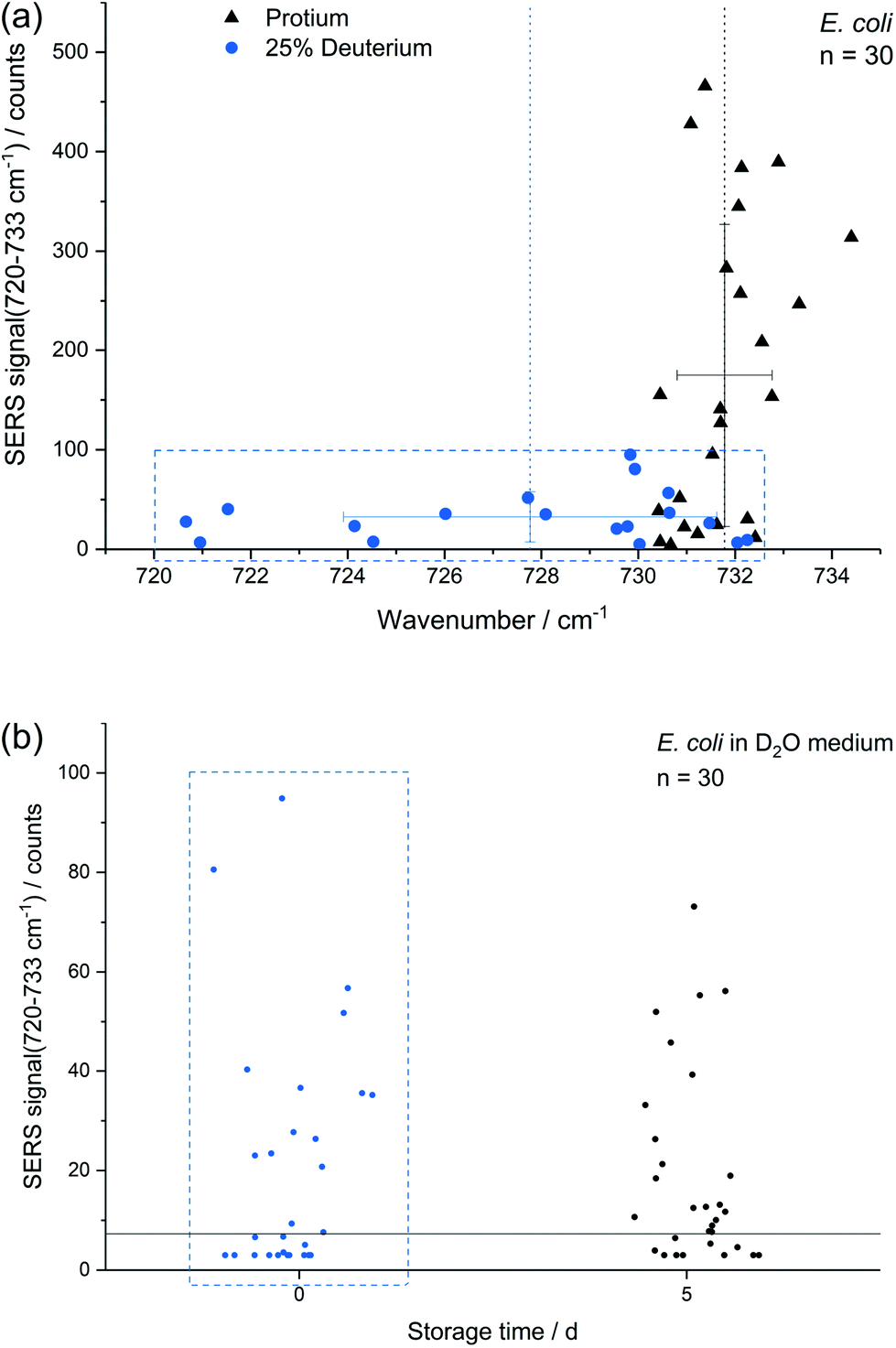 | ||
| Fig. 5 (a) SERS signal at around 733 cm−1 of E. coli with and without 25% D2O addition plotted against Raman shift (bars: 1 s); (b) SERS intensity of E. coli with 25% addition of D2O after 0 and 5 d storage. Note that the data at day 0 in the blue box of panel (b) corresponds to the deuterium data in the blue box of panel (a). | ||
The difference in SERS signal intensity and influence of storage on it between deuterated and unlabelled E. coli cells (Fig. 3a and 5b) underlines the impact of the physiological state of microbial target cells onto the results of SERS analyses and the necessity to conduct further research to explore how growth conditions and environmental factors affect microbial SERS signals in a species-dependent manner. On the other hand, our results suggest that SERS might be useful to analyse certain aspects of the physiology of E. coli with single-cell resolution, e.g. activity state of single cells or reaction upon stress conditions. Cells with distinct SERS signals likely are metabolic active.
The usefulness of spectroscopic techniques to detect active metabolic pathways or differentiate between certain physiological states was presented by Avetisyan et al. for Raman and by Muhamadali et al. for infrared spectroscopy.51,52 Stevenson et al. even demonstrated the detection of specific enzymatically catalysed reactions by SERS.53 The applicability of SERS onto environmental samples was shown by Cui et al. who analysed the incorporation of stable isotope-labelled nitrogen sources with microcosms from surface water.4 In the following sections we explore further opportunities for the applicability of SERS, in particular the strong signal at about 730 cm−1 in E. coli cells, for SERS-assisted sorting by optical tweezers and SERS for 3D imaging at the single-cell level.
Applicability of SERS for microbial detection
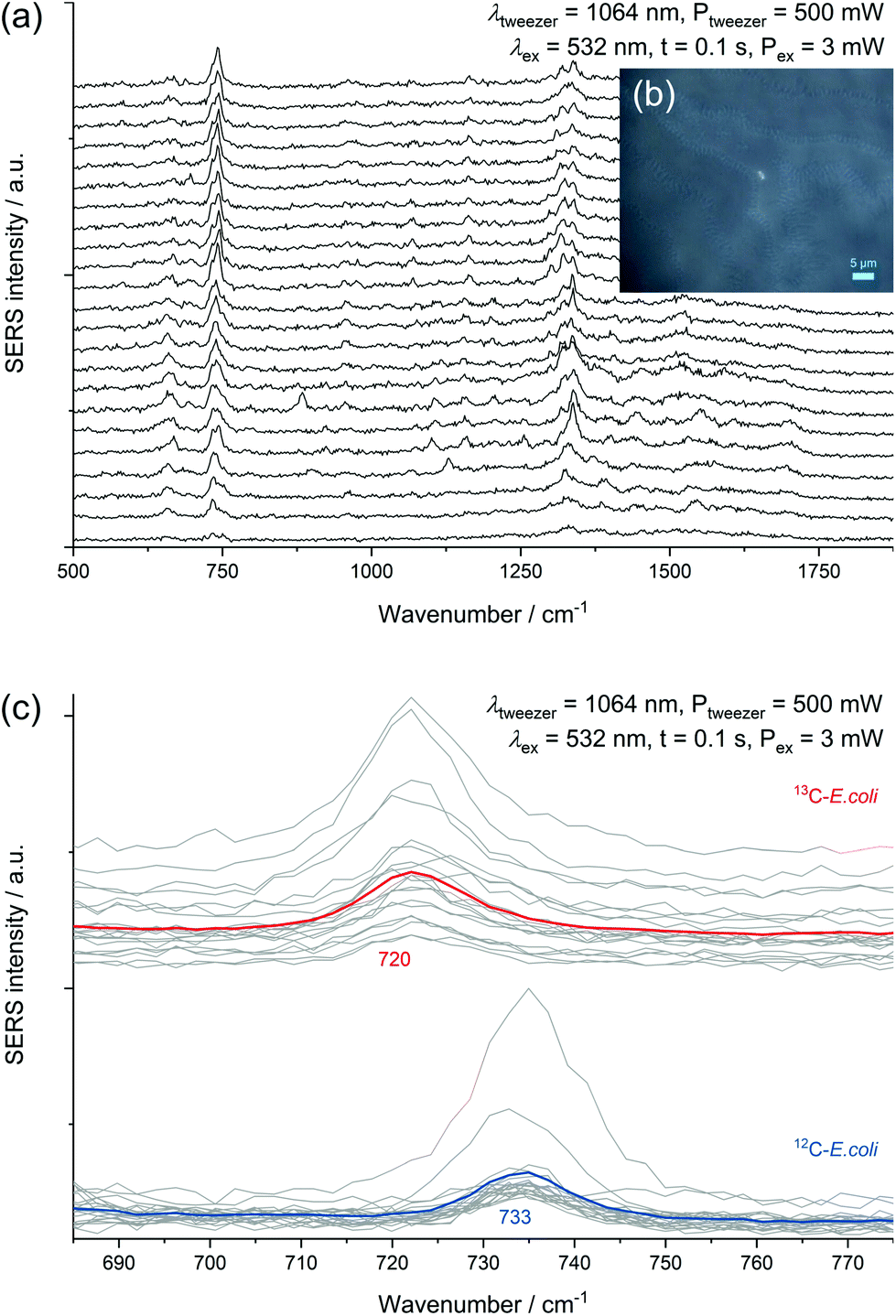 | ||
| Fig. 6 (a) Continuously acquired spectra of an AgNP@E. coli particle inside the laser focus. (b) Microscopic image during sorting by optical tweezing with an AgNP@E. coli particle inside the laser focus. (c) Consecutive SERS spectra of 13C-E. coli (red mean spectrum) and 12C-E. coli (blue mean spectrum) inside of the same sample with activated optical tweezer laser. Associated spectra are shifted on the ordinate for a better visualization. | ||
Experiments performed with a mixture of unlabelled and fully 13C-labelled E. coli proved that 13C-isotope incorporation into microbial cells can be measured by SERS of trapped cells (Fig. 6c). For two separate measurement events, we observed the normal SERS signal at 733 cm−1 with unlabelled E. coli cells and the red-shifted signal at 720 cm−1 for labelled cells. This detection of SERS signals of either fully labelled or unlabelled cells in a mixed sample combined with the optical evidence of separated cells suggest the applicability of SERS for sorting at the single-cell level. A general drawback of the experimental procedure is the necessity of an additional sample preparation step following in situ AgNP synthesis. The need for the removal of excess reagent implies difficulties to transfer the setup to more complex samples. The partial formation of larger aggregates because of the centrifugation calls for future optimization of the procedure. Especially the aforementioned discrepancy in SERS signals of different species represents a disadvantage for the use in sorting of unknown species.
3D SERS of microbial samples
The release of SERS-active metabolites implies also limitations for the use of SERS techniques for the spatially resolved imaging of microorganisms in environmental samples. Nevertheless, we tested the in situ AgNP synthesis as a way to accomplish also a three-dimensional detection of E. coli. Fig. 7 displays the 3D SERS image of a simplistic artificial biofilm model prepared with unlabelled and 13C-labelled E. coli cells which were washed before the in situ AgNP synthesis and afterwards mixed into the agarose gel.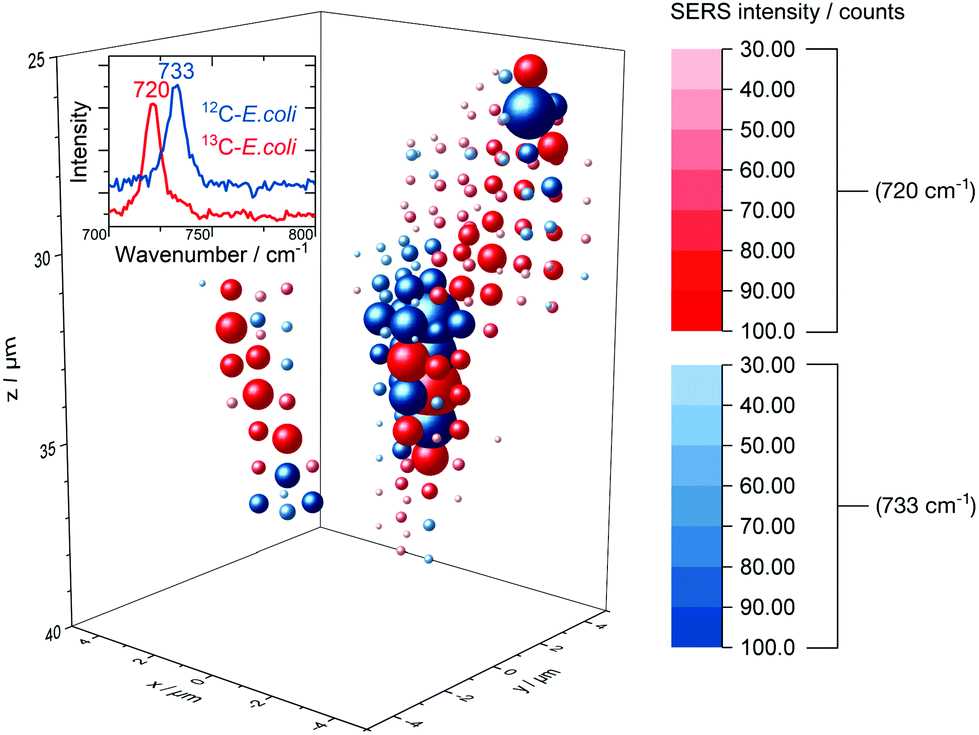 | ||
| Fig. 7 3D SERS image of 12C/13C-labelled E. coli cells embedded into an agarose matrix. Not shifted and red-shifted SERS signal are drawn at each grid position in blue and red spheres respectively, the size and saturation represent the intensity. The corresponding rotating iso-surface plot is available in the ESI.† | ||
Agarose gel is reported to be a suitable artificial model for biofilm research54 and therefore was used in this study. The distinct signal at around 730 cm−1 enables differentiation between labelled and unlabelled cells. The low integration time of 200 ms permits to scan volumes with grid intervals as depicted in Fig. 7 in about 30 min. Short measurements are required to precisely image aqueous biological samples as they are prone to misalign over time. Even though the SERS signal might be caused mainly by substances released from microbial cells we did not observe a broad dispersal of the SERS signal but found that it centred around cell structures. In addition, we tested the depth resolution of the Raman microspectroscopy setup with micrometre-sized synthetic polymer particles as shown in Fig. S10.† The results suggest that the depth resolution is sufficient to image micrometre-sized objects. Also, with a minimal ellipsoidal confocal volume of about 0.9 μm3 the grid interval of one micrometre is adequate to scan for SERS signals of E. coli cells, which own similar dimensions.
Conclusion
SERS spectra of six different microbial species (three Gram-negative, two Gram-positive and one archaeon) were analysed with identical measurement parameters giving rise to sharp peaks in case of E. coli but less distinct signals for the other species. The systematic comparison of fresh and stored E. coli cultures shows a rapid decrease of the SERS signal and the actual number of hits for single cells in stored cultures. This was the case both for bacteria cultured with a minimal medium as well as a full medium. The effect cannot be attributed to the death of cells since viability tests proved that after storage almost the same percentage of cells was still alive. SEM images showed that also in the case of stored cultures, i.e. cultures with lower SERS signal, AgNP were present on the cell surface eliminating detachment as another potential reason. We, therefore, relate the decline in SERS signal intensity to the metabolic activity of microbes and the release of nucleotides or their degradation products upon starvation and/or osmotic shock, e.g., adenine, guanine, hypoxanthine, xanthine as previously proposed.23 SERS spectra of partially deuterated bacteria (<25%) exhibit a red-shift of the characteristic SERS band at 733 cm−1 to 728 cm−1, a surprisingly larger variance of the peak position and a stable intensity of the SERS signal in samples stored up to 5 days in contrast to non-deuterated samples. Again, our conclusion is that the SERS intensity represents the starvation-induced release of substances related to the metabolic activity. We conclude that it is mandatory to take into account, and to conduct further research on, the effect of the physiological state of microbes onto the SERS signal of microbial samples. Otherwise an incomplete understanding may give rise to pitfalls in the characterization and evaluation, and to misinterpretations. Further, we stress the implication of metabolites as a potential cause of SERS signals. Since released substances are not restricted to the immediate vicinity of cells but diffuse out, spatial attribution may be compromised. In our work, washing of microbial cells prior to the in situ AgNP synthesis eliminated possible SERS contributions of metabolites already released into the culture medium. This conclusion is confirmed by three-dimensional analyses, which did not show a uniform distribution of SERS signals, as would be expected for metabolites in aqueous solution. On the contrary, a biofilm model with 12C- and 13C-labelled E. coli gave a telling visualization that SERS events were strictly confined to cellular structures. The same holds true for the sorting of E. coli cells by optical trapping via SERS-based detection: upon an additional centrifugation step we were able to tweeze microbial cells and attached AgNP without the contribution of background SERS signals. We conclude that – despite the observed variability of SERS signals which warrants further investigation – SERS has a potential for detection, sorting, and characterization of metabolically active microorganisms at the single-cell level.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support of the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG project IV 110/2-1), Federal Ministry of Education and Research (Bundesministerium für Bildung und Forschung, BMBF project 13N13698: LegioTyper) and TUM International Graduate School of Science and Engineering (IGSSE project 10.3 BIOMAG). MP and MW were supported by the European Research Council (Advanced Grant: NITRICARE 294343). We also would like to thank Fátima Pereira for the assistance with LTRS, Sina Stocker for essential contributions, Christine Sternkopf for the SEM measurements and Christoph Haisch for valuable discussions. We thankfully acknowledge helpful comments on the occurrence and effect of purines during microbial starvation and technical support from Markus Schmid.References
- J. Xu, J. W. Turner, M. Idso, S. V. Biryukov, L. Rognstad, H. Gong, V. L. Trainer, M. L. Wells, M. S. Strom and Q. Yu, Anal. Chem., 2013, 85, 2630–2637 CrossRef CAS PubMed.
- S. Polisetti, A. N. Bible, J. L. Morrell-Falvey and P. W. Bohn, Analyst, 2016, 141, 2175–2182 RSC.
- H. Guo, B. Xing, L. C. Hamlet, A. Chica and L. He, Sci. Total Environ., 2016, 554–555, 246–252 CrossRef CAS PubMed.
- L. Cui, K. Yang, G. Zhou, W. E. Huang and Y. G. Zhu, Anal. Chem., 2017, 89, 5793–5800 CrossRef CAS PubMed.
- X. Lu, D. R. Samuelson, Y. Xu, H. Zhang, S. Wang, B. A. Rasco, J. Xu and M. E. Konkel, Anal. Chem., 2013, 85, 2320–2327 CrossRef CAS PubMed.
- A. K. Boardman, W. S. Wong, W. R. Premasiri, L. D. Ziegler, J. C. Lee, M. Miljkovic, C. M. Klapperich, A. Sharon and A. F. Sauer-Budge, Anal. Chem., 2016, 88, 8026–8035 CrossRef CAS PubMed.
- F. Chen, B. R. Flaherty, C. E. Cohen, D. S. Peterson and Y. Zhao, Nanomedicine, 2016, 12, 1445–1451 CrossRef CAS PubMed.
- I. Marcaida, M. Maguregui, H. Morillas, C. Garcia-Florentino, V. Pintus, T. Aguayo, M. Campos-Vallette and J. M. Madariaga, Anal. Bioanal. Chem., 2017, 409, 2221–2228 CrossRef CAS PubMed.
- D. Berry, E. Mader, T. K. Lee, D. Woebken, Y. Wang, D. Zhu, M. Palatinszky, A. Schintlmeister, M. C. Schmid, B. T. Hanson, N. Shterzer, I. Mizrahi, I. Rauch, T. Decker, T. Bocklitz, J. Popp, C. M. Gibson, P. W. Fowler, W. E. Huang and M. Wagner, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E194–E203 CrossRef CAS PubMed.
- Y. Wang, W. E. Huang, L. Cui and M. Wagner, Curr. Opin. Biotechnol., 2016, 41, 34–42 CrossRef CAS PubMed.
- W. E. Huang, K. Stoecker, R. Griffiths, L. Newbold, H. Daims, A. S. Whiteley and M. Wagner, Environ. Microbiol., 2007, 9, 1878–1889 CrossRef CAS PubMed.
- E. Singer, M. Wagner and T. Woyke, ISME J., 2017, 11, 1949–1963 CrossRef CAS PubMed.
- C. Xie, D. Chen and Y.-Q. Li, Opt. Lett., 2005, 30, 1800–1802 CrossRef PubMed.
- W. E. Huang, A. D. Ward and A. S. Whiteley, Environ. Microbiol. Rep., 2009, 1, 44–49 CrossRef CAS PubMed.
- Y. Wang, Y. Ji, E. S. Wharfe, R. S. Meadows, P. March, R. Goodacre, J. Xu and W. E. Huang, Anal. Chem., 2013, 85, 10697–10701 CrossRef CAS PubMed.
- E. C. Le Ru, P. G. Etchegoin and M. Meyer, J. Chem. Phys., 2006, 125, 204701 CrossRef CAS PubMed.
- S. Efrima and B. V. Bronk, J. Phys. Chem. B, 1998, 102, 5947–5950 CrossRef CAS.
- R. M. Jarvis and R. Goodacre, Anal. Chem., 2004, 76, 40–47 CrossRef CAS PubMed.
- M. Kahraman, M. Müge Yazici, F. Şahİn, Ö. F. Bayrak, E. TopÇu and M. Çulha, Int. J. Environ. Anal. Chem., 2007, 87, 763–770 CrossRef CAS.
- P. Kubryk, J. S. Kolschbach, S. Marozava, T. Lueders, R. U. Meckenstock, R. Niessner and N. P. Ivleva, Anal. Chem., 2015, 87, 6622–6630 CrossRef CAS PubMed.
- L. Zeiri, B. V. Bronk, Y. Shabtai, J. Eichler and S. Efrima, Appl. Spectrosc., 2004, 58, 33–40 CrossRef CAS PubMed.
- P. Kubryk, R. Niessner and N. P. Ivleva, Analyst, 2016, 141, 2874–2878 RSC.
- W. R. Premasiri, J. C. Lee, A. Sauer-Budge, R. Theberge, C. E. Costello and L. D. Ziegler, Anal. Bioanal. Chem., 2016, 408, 4631–4647 CrossRef CAS PubMed.
- M. Chisanga, H. Muhamadali, R. Kimber and R. Goodacre, Faraday Discuss., 2017, 205, 331–343 RSC.
- H. Zhou, D. Yang, N. P. Ivleva, N. E. Mircescu, S. Schubert, R. Niessner, A. Wieser and C. Haisch, Anal. Chem., 2015, 87, 6553–6561 CrossRef CAS PubMed.
- H. Daims, E. V. Lebedeva, P. Pjevac, P. Han, C. Herbold, M. Albertsen, N. Jehmlich, M. Palatinszky, J. Vierheilig, A. Bulaev, R. H. Kirkegaard, M. von Bergen, T. Rattei, B. Bendinger, P. H. Nielsen and M. Wagner, Nature, 2015, 528, 504–509 CrossRef CAS PubMed.
- R. Hatzenpichler, E. V. Lebedeva, E. Spieck, K. Stoecker, A. Richter, H. Daims and M. Wagner, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2134–2139 CrossRef CAS PubMed.
- M. Palatinszky, C. Herbold, N. Jehmlich, M. Pogoda, P. Han, M. von Bergen, I. Lagkouvardos, S. M. Karst, A. Galushko, H. Koch, D. Berry, H. Daims and M. Wagner, Nature, 2015, 524, 105–108 CrossRef CAS PubMed.
- N. Leopold and B. Lendl, J. Phys. Chem. B, 2003, 107, 5723–5727 CrossRef CAS.
- H. Zhou, D. Yang, N. P. Ivleva, N. E. Mircescu, R. Niessner and C. Haisch, Anal. Chem., 2014, 86, 1525–1533 CrossRef CAS PubMed.
- L. Cui, S. Chen and K. Zhang, Spectrochim. Acta, Part A, 2015, 137, 1061–1066 CrossRef CAS PubMed.
- S. M. Beiser and B. D. Davis, J. Bacteriol., 1957, 74, 303–307 CAS.
- S. Efrima and L. Zeiri, J. Raman Spectrosc., 2009, 40, 277–288 CrossRef CAS.
- R. M. Jarvis and R. Goodacre, Chem. Soc. Rev., 2008, 37, 931–936 RSC.
- A. Walter, A. Marz, W. Schumacher, P. Rosch and J. Popp, Lab Chip, 2011, 11, 1013–1021 RSC.
- M. Kahraman, K. Keseroglu and M. Culha, Appl. Spectrosc., 2011, 65, 500–506 CrossRef CAS PubMed.
- A. Colnita, N. E. Dina, N. Leopold, D. C. Vodnar, D. Bogdan, S. A. Porav and L. David, Nanomaterials, 2017, 7, 248 CrossRef PubMed.
- H. Wetzel, B. Pettinger and U. Wenning, Chem. Phys. Lett., 1980, 75, 173–178 CrossRef CAS.
- R. M. Jarvis, A. Brooker and R. Goodacre, Anal. Chem., 2004, 76, 5198–5202 CrossRef CAS PubMed.
- Y. Liu, Y.-R. Chen, X. Nou and K. Chao, Appl. Spectrosc., 2007, 61, 824–831 CrossRef CAS PubMed.
- A. A. Guzelian, J. M. Sylvia, J. A. Janni, S. L. Clauson and K. M. Spencer, in Vibrational Spectroscopy-Based Sensor Systems, ed. S. D. Christesen and A. J. Sedlacek, SPIE—The International Society for Optical Engineering, Bellingham, 2002, vol. 4577, pp. 182–192 Search PubMed.
- S. W. Chiu, H. W. Cheng, Z. X. Chen, H. H. Wang, M. Y. Lai, J. K. Wang and Y. L. Wang, Phys. Chem. Chem. Phys., 2018, 20, 8032–8041 RSC.
- Y. Kim, C. M. Lew and J. D. Gralla, J. Bacteriol., 2006, 188, 7457–7463 CrossRef CAS PubMed.
- U. Rinas, K. Hellmuth, R. Kang, A. Seeger and H. Schlieker, Appl. Environ. Microbiol., 1995, 61, 4147–4151 CAS.
- M. E. Bayer, J. Bacteriol., 1967, 93, 1104–1112 CAS.
- M. Wagner, G. Rath, R. Amann, H.-P. Koops and K.-H. Schleifer, Syst. Appl. Microbiol., 1995, 18, 251–264 CrossRef CAS.
- E. Morgenroth, A. Obermayer, E. Arnold, A. Brühl, M. Wagner and P. A. Wilderer, Water Sci. Technol., 2000, 41, 105–113 CrossRef CAS.
- A. Bollmann, I. Schmidt, A. M. Saunders and M. H. Nicolaisen, Appl. Environ. Microbiol., 2005, 71, 1276–1282 CrossRef CAS PubMed.
- E. French and A. Bollmann, Life, 2015, 5, 1396–1404 CrossRef CAS PubMed.
- S. H. Kopf, S. E. McGlynn, A. Green-Saxena, Y. Guan, D. K. Newman and V. J. Orphan, Environ. Microbiol., 2015, 17, 2542–2556 CrossRef CAS.
- A. Avetisyan, J. B. Jensen and T. Huser, Anal. Chem., 2013, 85, 7264–7270 CrossRef CAS PubMed.
- H. Muhamadali, Y. Xu, D. I. Ellis, J. W. Allwood, N. J. Rattray, E. Correa, H. Alrabiah, J. R. Lloyd and R. Goodacre, Appl. Environ. Microbiol., 2015, 81, 3288–3298 CrossRef CAS PubMed.
- R. Stevenson, S. McAughtrie, L. Senior, R. J. Stokes, H. McGachy, L. Tetley, P. Nativo, J. M. Brewer, J. Alexander, K. Faulds and D. Graham, Analyst, 2013, 138, 6331–6336 RSC.
- M. Strathmann, T. Griebe and H. C. Flemming, Appl. Microbiol. Biotechnol., 2000, 54, 231–237 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional information as mentioned in the text. See DOI: 10.1039/c8an02177e |
| This journal is © The Royal Society of Chemistry 2019 |