
Dual cationic–anionic profiling of metabolites in a single identified cell in a live Xenopus laevis embryo by microprobe CE-ESI-MS†
 *a
*a
Abstract
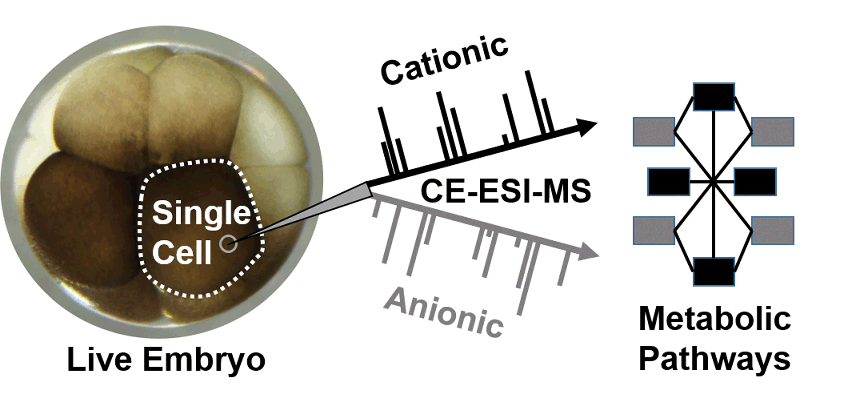
In situ capillary microsampling with capillary electrophoresis (CE) electrospray ionization (ESI) mass spectrometry (MS) enabled the characterization of cationic metabolites in single cells in complex tissues and organisms. For deeper coverage of the metabolome and metabolic networks, analytical approaches are needed that provide complementary detection for anionic metabolites, ideally using the same instrumentation. Described here is one such approach that enables sequential cationic and anionic (dual) analysis of metabolites in the same identified cell in a live vertebrate embryo. A calibrated volume was microaspirated from the animal-ventral cell in a live 8-cell embryo of Xenopus laevis, and cationic and anionic metabolites were one-pot microextracted from the aspirate, followed by CE-ESI-MS analysis of the same extract. A laboratory-built CE-ESI interface was reconfigured to enable dual cationic–anionic analysis with ∼5–10 nM (50–100 amol) lower limit of detection and a capability for quantification. To provide robust separation and efficient ion generation, the CE-ESI interface was enclosed in a nitrogen gas filled chamber, and the operational parameters were optimized for the cone-jet spraying regime in both the positive and negative ion mode. A total of ∼250 cationic and ∼200 anionic molecular features were detected from the cell between m/z 50–550, including 60 and 24 identified metabolites, respectively. With only 11 metabolites identified mutually, the duplexed approach yielded complementary information on metabolites produced in the cell, which in turn deepened network coverage for several metabolic pathways. With scalability to smaller cells and adaptability to other types of tissues and organisms, dual cationic–anionic detection with in situ microprobe CE-ESI-MS opens a door to better understand cell metabolism.
- This article is part of the themed collection: Next wave advances in single cell analyses
 Please wait while we load your content...
Please wait while we load your content...

