
Live single cell analysis using synchrotron FTIR microspectroscopy: development of a simple dynamic flow system for prolonged sample viability†

Abstract
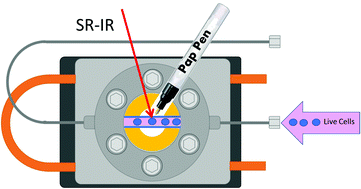
Synchrotron radiation Fourier transform infrared microspectroscopy (SR-microFTIR) of live biological cells has the potential to provide far greater biochemical and morphological detail than equivalent studies using dehydrated, chemically-fixed single cells. Attempts to measure live cells using microFTIR are complicated by the aqueous environment required and corresponding strong infrared absorbance by water. There is also the additional problem of the limited lifetime of the cells outside of their preferred culture environment. In this work, we outline simple, cost-effective modifications to a commercially available liquid sample holder to perform single live cell analysis under an IR microscope and demonstrate cell viability up to at least 24 hours. A study using this system in which live cells have been measured at increasing temperature has shown spectral changes in protein bands attributed to α–β transition, consistent with other published work, and proves the ability to simultaneously induce and measure biochemical changes. An additional study of deuterated palmitic acid (D31-PA) uptake at different timepoints has made use of over 200 individual IR spectra collected over ∼4 hours, taking advantage of the ability to maintain viable cell samples for longer periods of time in the measurement environment, and therefore acquire greatly increased numbers of spectra without compromising on spectral quality. Further developments of this system are planned to widen the range of possible experiments, and incorporate more complex studies, including drug–cell interaction.
- This article is part of the themed collection: Next wave advances in single cell analyses
 Please wait while we load your content...
Please wait while we load your content...

