
Analysing single live cells by scanning electrochemical microscopy
 *a
*a
Abstract
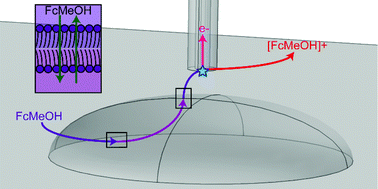
Single live cell analysis methods provide information on the characteristics of individual cells, yielding not only bulk population averages but also their heterogeneity. Scanning electrochemical microscopy (SECM) offers single live cell activities along its topography with high accuracy probe tip positioning. Both intracellular and extracellular processes can be electrochemically examined through the use of SECM. This non-invasive technique allows for high resolution mapping of electrochemical measurements in or around the cell sample of interest. Reactive oxygen species and reactive nitrogen species can be determined in a non-invasive label-free method and utilized as a probe for cellular pathology and physiology. Membrane permeability and rate of membrane species transport can be quantified in SECM. The cell response to external stressors can be monitored and modelled. SECM is able to offer nanoscale mapping and low concentration detection, providing a powerful bioanalytical tool for live cell studies. Herein we present an overview of recent progress in the imaging and characterization of single live cells using SECM.
- This article is part of the themed collections: Recent Review Articles and Next wave advances in single cell analyses
 Please wait while we load your content...
Please wait while we load your content...

