
Gel-based cell manipulation method for isolation and genotyping of single-adherent cells†
 b
Tomoko
Yoshino
*a
b
Tomoko
Yoshino
*a
Abstract
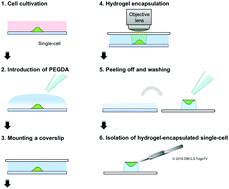
Genetic analysis of single-cells is widely recognized as a powerful tool for understanding cellular heterogeneity and obtaining genetic information from rare populations. Recently, many kinds of single-cell isolation systems have been developed to facilitate single-cell genetic analysis. However, these systems mainly target non-adherent cells or cells in a cell suspension. Thus, it is still challenging to isolate single-adherent cells of interest from a culture dish using a microscope. We had previously developed a single-cell isolation technique termed “gel-based cell manipulation” (GCM). In GCM, single-cells could be visualized by photopolymerizable-hydrogel encapsulation that made it easier to isolate the single-cells. In this study, GCM-based isolation of single-adherent cancer cells from a culture dish was demonstrated. Single-adherent cells were encapsulated in a photopolymerizable hydrogel using a microscope and isolated with high efficiency. Furthermore, whole genome amplification and sequencing for the isolated single-adherent cell could be achieved. We propose that the GCM-based approach demonstrated in this study has the potential for efficient analysis of single-adherent cells at the genetic level.
- This article is part of the themed collection: Next wave advances in single cell analyses
 Please wait while we load your content...
Please wait while we load your content...

