
Single-cell mobility shift electrophoresis reports protein localization to the cell membrane†

Abstract
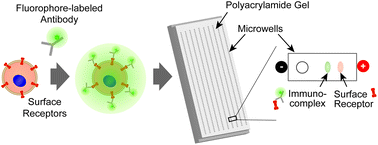
While profiling of cell surface receptors grants valuable insight on cell phenotype, surface receptors alone cannot fully describe activated downstream signaling pathways, detect internalized receptor activity, or indicate constitutively active signaling in subcellular compartments. To measure surface-bound and intracellular targets in the same cell, we introduce a tandem single-cell assay that combines immunofluorescence of surface-bound epithelial cellular adhesion molecule (EpCAM) with subsequent protein polyacrylamide gel electrophoresis (PAGE) of unfixed MCF7 breast cancer cells. After surface staining and cell lysis, surface EpCAM is analyzed by single-cell PAGE, concurrent with immunoprobing of intracellular targets. Consequently, the single-cell electrophoresis step reports localization of both surface and intracellular targets. Unbound intracellular EpCAM is readily resolved from surface EpCAM immunocomplex owing to a ∼30% mobility shift. Flow cytometry and immunofluorescence are in concordance with single-cell PAGE. Lastly, we challenged the stability of the EpCAM immunocomplexes by varying ionic and non-ionic component concentrations in the lysis buffer, the lysis time, and electrophoresis duration. As expected, the harsher conditions proved most disruptive to the immunocomplexes. The compatibility of live-cell immunostaining with single-cell PAGE eliminates the need to perform single-cell imaging by condensing read-out of both surface-bound proteins (as low mobility immune complexes) and intracellular targets to a single immunoblot, thus linking cell type and state.
- This article is part of the themed collection: Next wave advances in single cell analyses
 Please wait while we load your content...
Please wait while we load your content...

