
The in vivo effects of silver nanoparticles on terrestrial isopods, Porcellio scaber, depend on a dynamic interplay between shape, size and nanoparticle dissolution properties†
Sara
Novak
*a,
Tea
Romih‡
a,
Barbara
Drašler§
a,
Giovanni
Birarda
 b,
Lisa
Vaccari
b,
Paolo
Ferraris¶
b,
Stephanie
Sorieul
c,
Maciej
Zieba
de,
Victor
Sebastian
de,
Manuel
Arruebo
de,
Samo B.
Hočevar
f,
Anita
Jemec Kokalj
b and
Damjana
Drobne
c
b,
Lisa
Vaccari
b,
Paolo
Ferraris¶
b,
Stephanie
Sorieul
c,
Maciej
Zieba
de,
Victor
Sebastian
de,
Manuel
Arruebo
de,
Samo B.
Hočevar
f,
Anita
Jemec Kokalj
b and
Damjana
Drobne
c
aDepartment of Biology, Biotechnical Faculty, University of Ljubljana, Ljubljana, Slovenia
bElettra - Sincrotrone Trieste SCpA, AREA Science Park, Basovizza, Trieste, Italy
cUniversity of Bordeaux, CENBG, UMR 5797, Gradignan Cedex, France. E-mail: damjana.drobne@bf.uni-lj.si
dInstitute of Nanoscience of Aragon (INA) and Department of Chemical and Environmental Engineering, University of Zaragoza, Spain
eNetworking Research Center on Bioengineering, Biomaterials and Nanomedicine, CIBER-BBN, 28029-Madrid, Spain
fDepartment of Analytical Chemistry laboratory, National Institute of Chemistry, Ljubljana, Slovenia
First published on 19th October 2018
Abstract
The present work aims to study the effects that acute exposure to low concentrations of silver nanoparticles (AgNPs) cause in digestive glands of terrestrial isopods (Porcellio scaber). The experiments were designed to integrate different analytical techniques, such as transmission electron microscopy, atomic absorption spectroscopy, proton induced X-ray emission, and Fourier transform IR imaging (FTIRI), in order to gain a comprehensive insight into the process from the AgNPs’ synthesis to their interaction with biological tissues in vivo. To this aim, terrestrial isopods were fed with AgNPs having different shapes, sizes, and concentrations. For all the tested conditions, no toxicity at the whole organism level was observed after 14 days of exposure. However, FTIRI showed that AgNPs caused detectable local changes in proteins, lipids, nucleic acids and carbohydrates at the tissue level, to an extent dependent on the interplay of the AgNPs’ properties: shape, size, concentration and dissolution of ions from them.
Introduction
Silver nanoparticles (AgNPs) are one of the most commonly used nanomaterials, applied in more than several hundred commercial products1 and their applications are continuing to increase.2–4 The predicted environmental concentrations of AgNPs in sediments (in the U.S.) range between 0.7 and 2.2 μg kg−1.5An examination of the existing literature on AgNP toxicity reveals that it is still unclear which of the AgNPs’ properties plays the predominant role in determining their adverse effects. It is generally accepted that the release of Ag ions from AgNPs is an important factor in determining the observed toxic effects.6–8 However, a number of authors have observed that the toxic effects cannot be explained solely as a response to dissolved ions,6,9–11 and that NP size and shape also modulate their biological effects.12,13 It has been reported that the size of AgNPs dictates their bio-distribution and toxicity in mice,14 zebrafish,15 bacteria,16 yeast,11 and in vitro on different cell lines,17,18 such as mammalian white blood cell lines.19 A number of authors also reported a correlation between NP shape and toxicity, e.g. for bacteria,20,21 plants,10,22 and fishes.23 However, Gorka and coworkers10 could not confirm the correlation between AgNP shape and the effects on model organisms (Danio rerio and Caenorhabditis elegans) and bacterial species (Esherichia coli, Bacillus cereus, and Pseudomonas aeruginosa), and Park and Yeo17 suggested that diverse shapes of nanosilver (nanocolloids and nanotubes) caused similar patterns of metabolic responses in zebrafish embryos.
The aim of our study was to investigate the interplay between the intrinsic properties of AgNPs (size, shape, concentration and dissolution potential) in determining their effects on a model organism (Porcellio scaber, isopoda, Crustaceae) after the ingestion at subtoxic concentrations. To this end, the test organisms were fed with AgNPs at different subtoxic concentrations, having different size and shapes: Ag cubical NPs (NCs) with an average size of 60 nm, spherical NPs with an average particle size of 5–6 nm (NSs 5–6 nm) and spherical particles with average particle sizes of 11–12 nm (NSs 11–12 nm). AgNP shape, size, dispersibility and peculiar dissolution properties were evaluated, exploiting several analytical methods. The integrated organism response to the administration of the diverse NPs at different concentrations was also investigated by studying Ag ion internalization in isopod digestive glands. Finally, Fourier transform infrared imaging (FTIRI) was used to compare the spectral patterns of the digestive glands of non-exposed animals and those exposed to different types of AgNPs and to link these data to Ag assimilated by digestive gland cells and to conventional toxicological parameters.
Materials and methods
Chemicals
Silver nitrate (AgNO3, 99.9999 wt% trace metal basis), polyvinylpyrrolidone (PVP, Mw = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da and 55000 Da), ethylene glycol (EG, anhydrous, 99.8 wt%) and sodium hydrosulfate hydrate (≥60 wt%) were purchased from Sigma–Aldrich and used as received. Milli-Q grade water and acetone (grade ≥99.5%) were used as solvents.
000 Da and 55000 Da), ethylene glycol (EG, anhydrous, 99.8 wt%) and sodium hydrosulfate hydrate (≥60 wt%) were purchased from Sigma–Aldrich and used as received. Milli-Q grade water and acetone (grade ≥99.5%) were used as solvents.
AgNP synthesis
Ag spherical nanoparticles (AgNSs) were prepared following the work of Komarneni and co-workers24 with slight variations to produce spherical nanoparticles 5–6 nm and 11–12 nm in diameter, AgNS_5–6 and AgNS_11–12 hereafter. Silver nanocubes (AgNCs) were prepared using an experimental process adapted from the polyol synthesis of NCs reported by Siekkinen and collaborators.25 For both syntheses, PVP was used for preventing particle aggregation by PVP coating. More details on the AgNP synthesis are reported in the ESI.†AgNP characterization
The morphology of the nanoparticles was investigated by transmission electron microscopy (TEM) using a FEI Tecnai T20 microscope, operating at 200 kV. Briefly, 50 μL of a colloidal AgNP dispersion in water were deposited onto a carbon-coated copper grid and dried at room temperature. After the solvent was evaporated, the grid was observed under a TEM microscope in the bright field mode. UV–Vis spectra of the colloidal AgNP dispersions in water were obtained with a PerkinElmer Lambda 2 spectrophotometer, at a rate of 60 nm min−1 and a spectral resolution of 2 nm, using a quartz cell with a pathlength of 1 cm. The blank was the corresponding water solution.AgNPs were only characterised in the stock dispersion or the powder applied onto the leaves, but not in the actual leaves offered to the animals. Namely, the analysis of AgNP size and number in complex biological matrices is still not without methodological constraints.26 Namely, false positive formation of Ag NPs from Ag+ can be observed. Additionally, in our case the AgNPs are additionally transformed inside the guts of animals; therefore, the characterisation of AgNPs on the leaves does not contribute to the interpretation of data.
AgNP dissolution measurements
AgNP stock solutions were prepared in deionized water at the following nominal concentrations: 50 mg L−1 of AgNP for AgNC and AgNS 11–12 nm, and 43 mg L−1 of AgNS for AgNS_5–6 nm. The actual concentration of total Ag in diluted AgNP suspensions of the stock solutions was checked with flame AAS after overnight digestion in 1 M HNO3 (suspension/acid ratio 1:1 vol/vol).
Furthermore, eight milliliters of the stock AgNP suspension were ultracentrifuged at 37500 rpm (100000g) for 30 min at 20 °C (Beckman Coulter L8–70 M class H preparative ultracentrifuge with Type 70.1 Ti rotor and 10 mL thick-wall polyallomer tubes). The supernatants were divided into two aliquots. The total Ag concentrations in the first series of supernatant aliquots were determined by flame Atomic Absorption Spectroscopy, AAS (PerkinElmer AAnalyst 100, Waltham, Massachusetts, USA) after an overnight digestion in 10% (vol/vol) aqua regia. The second series of supernatant aliquots was left un-acidified and was analyzed by anodic stripping voltammetry (ASV) at the bismuth film electrode (BiFE) following the protocol described by Romih et al.27 The free Ag(I) ions were determined by the method of three standard additions, and their concentrations were calculated by linear regression. The Ag ion concentrations in supernatants obtained by ASV were compared to the total Ag concentrations in the corresponding stock suspensions and the percentages of dissolved Ag ions were calculated.
Design of the experiment
Terrestrial isopods (Porcellio scaber, Isopoda, Crustaceae) were collected in June 2012 at an noncontaminated location near Ljubljana, Slovenia. The animals were kept in the laboratory as described in our previously published studies.28 Only adult insects of both sexes, which did not molt and did not have marsupia, and weighing more than 25 mg were selected. Dried hazelnut leaves (Corylus avellana) weighing 100 ± 10 mg were used as a food source onto which AgNP suspensions in water were applied. The application was done according to our established procedure.27 This procedure has previously proved to result in final concentrations of NPs that were close to nominal.27 100 μL of a test chemical per 100 mg of leaf was evenly distributed onto abaxial leaf surfaces using a brush. The total Ag concentrations that were applied on the leaves were: 3.6 and 36 μg Ag per mL for AgNC, 0.3 and 3 μg Ag per mL for AgNS_5–6 and 0.28 and 2.8 μg Ag per mL for AgNS_11–12. This resulted in the following nominal concentrations of Ag: 3.6 and 36 μg Ag per g of leaf, 0.3 and 3 μg Ag per g of leaf, and 0.28 and 2.8 μg Ag per g of leaf, for AgNC, AgNS_5–6 and AgNS_11–12, respectively. In the control group, the leaves were spiked with deionized water. The leaves were allowed to dry for 24 h at room temperature and weighed. The leaves were used after 24 h of drying. Each individual insect was placed on a 9 cm Petri dish with one treated leaf as the only food supply. The food was not changed during the experiment. The experiment was carried out for 14 days. Faeces were removed on the 7th day of the experiment. All Petri dishes were kept in a large glass container under controlled conditions in terms of air humidity (≥80%, maintained by spraying tap water on the internal side of the lid every day), temperature (21 ± 1 °C) and light regime (16:8 h light:dark photoperiod).
We conducted three 14 days feeding experiments; only the number of exposed animals varied among the experiments, depending on the analyses to be carried out after NPs’ exposure. For the further analyses the insects were anaesthetized at low temperature, decapitated and their digestive glands were isolated. After exposure, faecal pellets and leaves were removed from the Petri dishes, dried at room temperature for 24 h and weighed. The feeding rate of the isopods was calculated as the mass of consumed leaves per wet fresh weight of the animal body mass per day. Food assimilation efficiency was calculated as the difference between the mass of consumed leaves and the mass of faecal pellets divided by the mass of consumed leaves.
Ag concentrations in whole digestive glands
After the 14-day exposure period, the animals from one of the feeding experiments were transferred to new Petri dishes and fed with uncontaminated hazel leaves for 24 h to remove any remaining Ag from their digestive system. The total Ag concentrations in the isolated digestive glands of isopods were determined by flame AAS. Prior to the analysis, the samples were acid digested in concentrated nitric acid (65% HNO3, analytical grade purity, Fisher Scientific, Loughborough, UK) in a Milestone Ethos E microwave lab station (Milestone, Bergamo, Italy) equipped with a SK-10 high pressure segmented rotor and 3 mL quartz microsampling inserts. Digestion was conducted at 180 °C and 600 W power, with step 1 (heating) lasting 15 min, step 2 (constant temperature at 180 °C) lasting 10 min, and 45 min of cooling to 60 °C.Elemental analyses of digestive gland cross sections
Particle induced X-ray emission (PIXE) analysis was used to study the internalization of silver in the digestive gland cells after the completion of each feeding experiment. PIXE measurements were performed at the AIFIRA facility (CENBG, Gradignan, France) as described by Sorieul et al.29 on digestive gland cryosections, 60 microns thick, mounted between two thin layers of Pioloform foil. A 3 MeV proton microbeam (size = 2.5 μm, beam current = 1 nA) was used in order to carry out the 2D elementary mapping of the samples. The use of a filter mounted on a Si(Li) detector guaranteed a dead time below 10%. An elementary quantification was performed using MicroMatter standards. Data treatment was carried out using GUPIX software. We analyzed 2 controls, 3 samples of animals exposed to AgNC, and 2 samples of animals exposed to each type of AgNS at both tested concentrations.FTIR imaging of digestive gland cell cross sections
Samples from 10 exposed animals were prepared for FTIR analyses as 15 μm-thin cryo-sections using a Leica CM3050 cryotome and the Jung Tissue Freezing Medium, Leica. The sections were then placed onto 2 mm-thick CaF2 IR transparent windows, which were transferred to a freeze dryer (Alpha 2–4 Christ) using a cryo-transfer assembly cooled with liquid nitrogen, and then freeze dried at −30 °C and 0.4 mbar for 24 h. Three different animals were investigated for control and for the groups exposed to AgNS_5–6-dosed food 2.8 μg Ag per g leaf and to AgNS_11–12-dosed food at both concentrations. Two animals were studied for the groups exposed to AgNS_5–6-dosed food 0.28 μg g−1 leaf and to AgNC 3.6 μg g−1 leaf. Finally, in the group exposed to AgNC-dosed food 36 μg g−1 leaf, four animals were examined. The samples were measured at the SISSI beamline (Synchrotron Infrared Source for Spectroscopy and Imaging), the infrared beamline of Elettra Sincrotrone Trieste30 with conventional Glow-bar source, using the 64 × 64 element Focal Plane Array detector equipped with the Hyperion 3000 IR/VIS microscope coupled with a Vertex70 interferometer (Bruker Optics GmbH – Ettlingen). Chemical images of the samples were obtained in transmission mode by averaging 256 scans with a spectral resolution of 4 cm−1. Using a 15× condenser/objective with 0.4 NA, the pixel resolution of the images is about 2.6 × 2.6 microns. Obtained hyperspectral images were corrected for environmental spectral contributions of water vapor and CO2, using the atmospheric compensation routine of OPUS 7.5 (Bruker Optics GmbH – Ettlingen). The files were then converted into ENVI format and processed in R using HyperSpec, baseline corrected (fillPeaks method), and processed with Rclusterpp packages.31–33 Image pixels containing the embedding medium were removed using an in-house written procedure based on the intensity of the 2945 cm−1 band in the second derivative spectra.34 Moreover, spectra with a peak height of 1660 cm−1 smaller than 0.1 a.u. and higher than 1.5 a.u. were discarded. A first data set was made grouping all the pre-processed spectra (∼133.000 spectra), disregarding the sample location while retaining information on the treatment. Both univariate analysis and Principal Component Analysis (PCA) were applied on standard vector normalized files. Intensity profiles of the main macromolecules were calculated as area integrals. The following spectral ranges were considered: for proteins the 1700–1480 cm−1 spectral region of amide I and II (Region 1); for lipids and, to a less extent, aliphatic chains of aminoacids, the symmetric and asymmetric stretching of –CH2 and –CH3 chemical moieties at 3100–2800 cm−1 (Region 2); and for nucleic acids the 1260–1190 cm−1 spectral region, representative of the asymmetric stretching phosphate moieties in nucleic acids and phospholipids (Region 3). PCA analysis on the entire data set was done on pre-processed standard vector normalized absorbance spectra, considering the following region of interest, ROI: 3050–2800 & 1780–910 cm−1.A second PCA analysis was run on the chemical images, using the same parameters. To this aim, all the images were processed together, retaining location information beyond the treatment, and PCA done in a single run.
Results
Nanoparticle characteristics
AgNPs were synthesized in different shapes and dimensions following the protocols detailed in the ESI.† TEM images shown in Fig. 1 (top and middle panels) reveal the morphology of AgNPs used for the feeding experiments: 60 nm nanocubes with a truncated cubic shape (AgNC), spherical NPs with primary particle sizes from 5 to 6 nm (AgNS_5–6), and spherical NPs with primary particle sizes from 11 to 12 nm (AgNS_11–12). Particle size distribution was calculated using the TEM images, averaging at least 50 particles.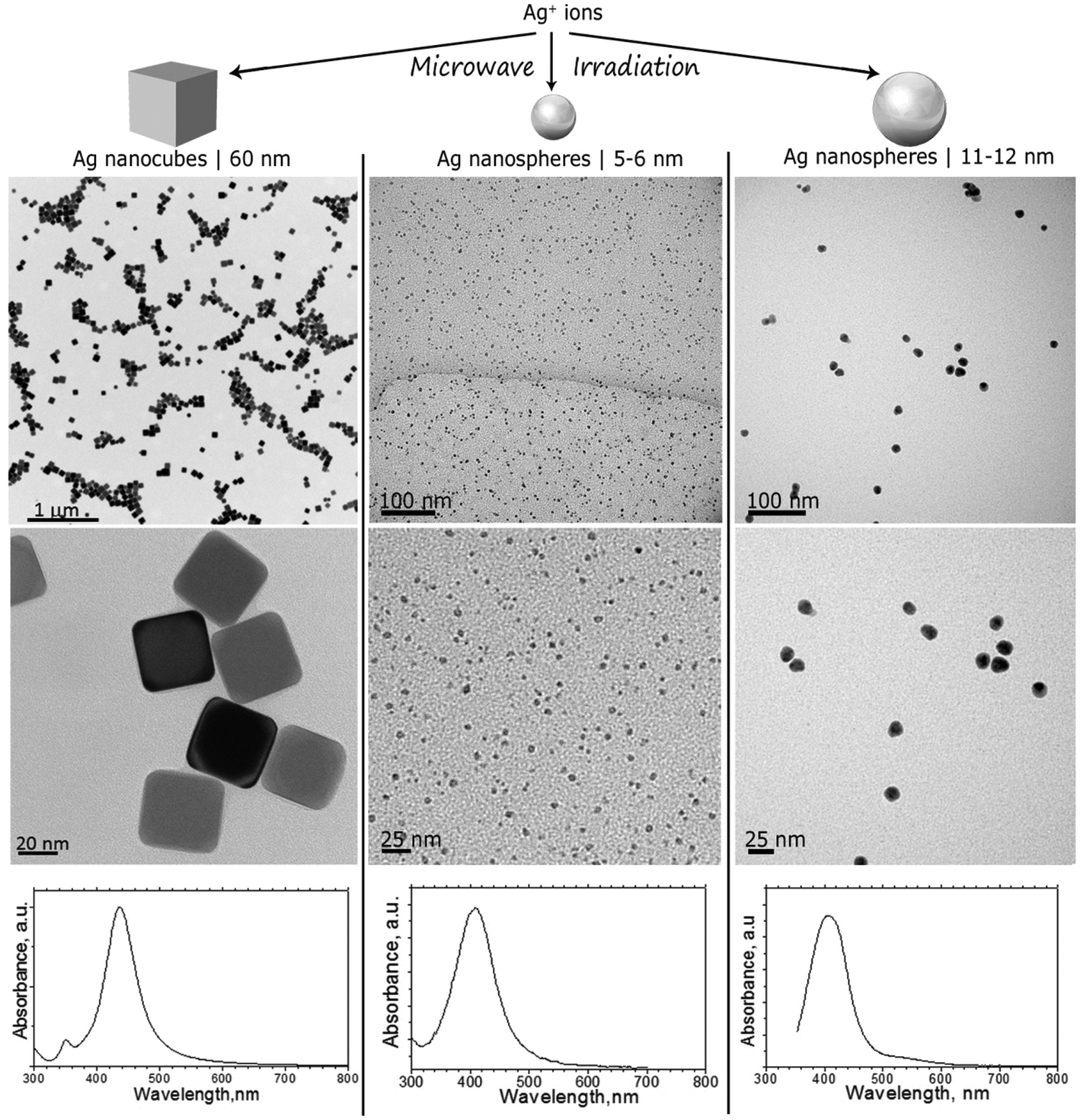 | ||
| Fig. 1 TEM images and UV-Vis spectra of AgNCs with sizes around 60 nm, AgNSs with sizes ranging between 5 and 6 nm (AgNS_5–6), and AgNSs with particle sizes ranging between 11 and 12 nm (AgNS_11–12). | ||
TEM images indicated well dispersed NPs, as also confirmed by UV-Vis spectrophotometry. As can be seen in Fig. 1 (bottom panels), characteristic localized surface plasmon resonances, LSPRs, were centered at 404 nm for all investigated NPs, with a reduced full width at half maximum, which corroborates a reduced polydispersity indicative of successful stabilization provided by the PVP grafted on the surface of the nanostructures. Indeed, the Mie theory calculations of the extinction spectra of silver nanoparticles also predict localized SPR peaks at around 410 nm.35
Stock suspensions of the synthesized AgNPs for feeding experiments were prepared in deionized water, and the total silver contained was determined by flame AAS (see Total Ag_stock in Table 1). ASS was also employed for determining the total Ag concentration in the supernatant of the stock dispersion after ultracentrifugation (Total Ag_sup). The obtained values, reported in Table 1, differ from the results obtained by ASV, that gives the concentration of free Ag(I) ions (Ag+_sup). The discrepancy can be ascribed to the incomplete sedimentation of AgNPs under the employed ultracentrifugation conditions, as already reported by Romih et al.36 The dissolved concentrations of Ag+ ions determined by ASV was therefore considered in order to estimate the percentage of dissolved silver for each stock AgNP suspension (%Ag+_sup). The obtained results, summarized in Table 1, testify that approximately the same percentage of silver, ranging between 1 and 2%, dissolves from all the tested AgNPs for any of the tested AgNP suspensions.
| AgNP type | Total Ag_stock | Total Ag_sup | Ag+_sup | Ag+_sup |
|---|---|---|---|---|
| n = 2 | n = 2 | n = 1 | ||
| [mg L−1] | [mg L−1] | [mg L−1] | [%] | |
| AgNC | 38.4 ± 1.1 | 1.4 ± 0.3 | 0.83 | 2.16 |
| AgN_5–6 | 50.8 ± 0.15 | 1.1 ± 0.2 | 1.02 | 2.02 |
| AgNS_11–12 | 46.3 ± 1.2 | 0.9 ± 0.1 | 0.58 | 1.3 |
Changes in feeding parameters of terrestrial isopods
For the purpose of these experiments, Porcellio scaber feeding experiments were done in triplicate where the animals were exposed for 14 days to leaves dosed with AgNPs, providing nominal concentrations of 3.6 and 36 μg Ag per g of leaf, 0.3 and 3 μg Ag per g of leaf, and 0.28 and 2.8 μg Ag per g of leaf, for AgNCs, AgNS_5–6 and AgNS_11–12 respectively. The weight change of the animals, their feeding rate, and food assimilation efficiency were not affected at these oral exposure concentrations for any of the tested AgNPs. Also, no mortality of animals during the experiments was noticed (see Fig. S1–S3 in ESI†).Ag concentration and distribution in digestive gland epithelium
Despite the fact that we could not detect any adverse effect at whole organism level after 14 days of exposure to AgNPs, the amount of Ag was significantly increased in the digestive glands of P. scaber in all exposed groups in comparison to controls. Indeed, digestive gland epithelium, also known as hepatopancreas, is composed of four blind-ending tubes within the body cavity and provides to animals both digestive and absorption function. As can be seen in Fig. 2, Ag accumulation was more pronounced for the groups of animals exposed to AgNCs at both concentrations (3.6 and 36 μg Ag per g of leaf) and to the highest tested concentrations of AgNSs, 3 and 2.8 μg Ag per g of leaf for AgNS_5–6 and for AgNS_11–12 respectively. Moreover, considering NPs with the same spherical shape, Ag from smaller AgNSs tends to accumulate more in digestive gland epithelium with respect to larger AgNS_11–12.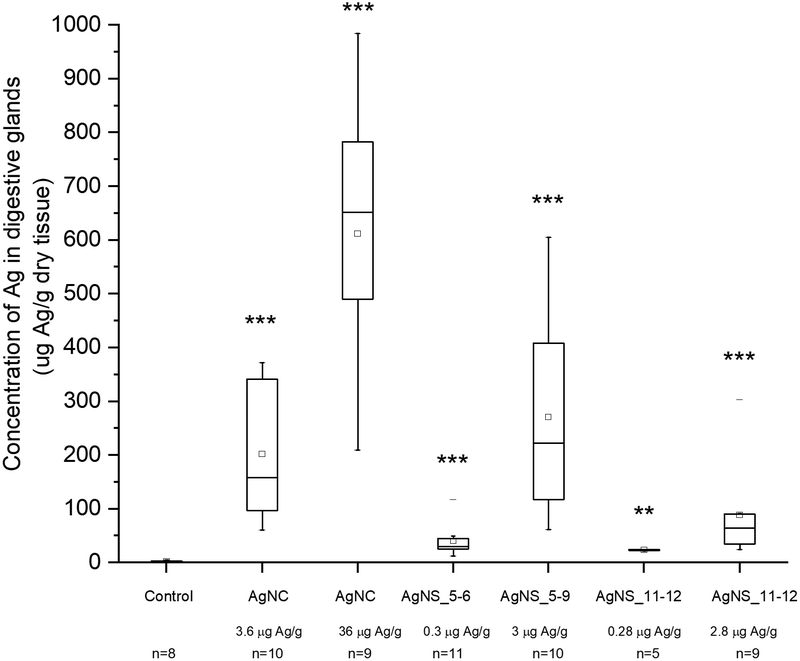 | ||
| Fig. 2 Concentration of Ag in the hepatopancreas after 14 days of feeding with AgNP-dosed food, as assessed by flame AAS. Symbols on the box plot represent minimum and maximum data values (whiskers), mean value (□), 75th percentile (upper edge of box), 25th percentile (lower edge of box), median (line in box) and max and min value ( – ). Statistical differences between the exposed and the control animals are marked with * (p < 0.05* and p < 0.01**). Exposure concentrations and the number of animals (n) per group are provided on the x-axis. | ||
Micro-PIXE analysis on a digestive gland cross section was done in order to highlight the Ag cellular distribution. Specifically, the digestive gland tube contains two cell types, large B-cells, that exert secretory and absorptive function, and wedge-shaped S-cells, (see Fig. 3, panel A). S-cells accumulate large amounts of metals, usually in spherical metalliferous granules containing mostly Cu, S and Ca.37 Therefore, the distribution of Cu was determined as well in order to locate S-cells of the digestive gland epithelium and metal storing granules within them. In Fig. 3, panels b and c, the distribution of Cu and Ag is shown for a section of the digestive gland tube of an ispodod exposed to AgNC-dosed food with at an exposure concentration of 36 μg Ag per g per leaf. In all the samples, the Ag signal overlaps with the Cu one. The results indicate that Ag is stored in the metal storing granules of S-cells.
 | ||
| Fig. 3 PIXE analyses of digestive gland cross-sections. a. Representative visible image of isopod digestive gland cross-section. The sections are taken from the different parts of the digestive gland as the sections of the tubes from panel b and c. b, c Elemental maps of Cu and Ag distribution into two cross sections of digestive glands of animals exposed to AgNC-dosed food at an exposure concentration of 36 μg Ag per g of leaf. | ||
A quantitative analysis on PIXE images was also performed, aimed to determine the concentration of Ag and Cu on digestive gland cross-sections. Data reported in Table 2 show that the main factor driving the accumulation of Ag is AgNP concentration, since the highest levels of accumulated Ag were found in animals exposed to the highest AgNC concentration tested (36 μg Ag per g of leaf). Concentration dependent difference in accumulated Ag was also found between the lower and higher concentrations of the same type of Ag nanomaterials. For example, an evidently higher amount of Ag was accumulated in the case of 2.8 μg Ag per g of AgNSs in comparison to 0.28 μg Ag per g of the same material (sample AgNS_5–6), and in the case of 36 μg Ag per g of AgNCs in comparison to ten-fold lower levels of AgNCs. However, the same trend was not observed for another specimen exposed to AgNCs (sample AgNS_11–12) which indicates high inter-individual variability. This is in line with variability observed also in other parameters, such as concentration of Ag in the hepatopancreas (Fig. 2). In addition, the higher accumulation potential of AgNS_5–6 with respect to AgNS_11–12 can be deduced by the data reported in Table 2. Overall, the PIXE results are in accordance with flame AAS data, and complement them by giving information on Ag localization.
| Sample | Concentration | |
|---|---|---|
| Ag (ng cm−2) | Cu (ng cm−2) | |
| Control A | <LOD | 1465.8 ± 4.2 |
| Control B | <LOD | 1044.9 ± 3.3 |
| AgNC | 308.3 ± 10.8 | 1565.1 ± 3.4 |
| 3.6 μg Ag per g | ||
| AgNC | 1050.1 ± 11.13 | 1378.8 ± 1.93 |
| 36 μg Ag per g | ||
| AgNS_5–6 | <LOD | 487.1 ± 1.7 |
| 0.3 μg Ag per g | ||
| AgNS_5–6 | 415.8 ± 12.1 | 1229.5 ± 3.1 |
| 3 μg Ag per g | ||
| AgNS_11–12 | 102.3 ± 6.6 | 993.3 ± 2.6 |
| 0.28 μg Ag per g, A | ||
| AgNS_11–12 | 345.7 ± 18.1 | 6464.5 ± 10.3 |
| 0.28 μg Ag per g, B | ||
| AgNS_11–12 | 226.8 ± 14.2 | 3814.8 ± 7.3 |
| 2.8 μg Ag per g | ||
Analyses of molecular changes in digestive gland cell cross sections by FTIR imaging
For the purpose of the analysis, all FTIR microspectra, pre-processed as described in the Materials and Methods, were grouped together and analysed. Spectra of each exposure group were analysed independently from their spatial localization. The results of PCA analysis are shown in Fig. 4. For clarity reasons, due to the large number of input data, the PC2 vs. PC1 score plot has been divided into three partial plots, each of them containing the control animal and the AgNCs, AgNS_5–6 and AgNS_11–12 animals in panels a, b and c respectively. The plots reveal that principal components 1 and 2 account for about 75% of the variance of the dataset: PC1 48%, PC2 26%. PC2 vs. PC1 space in Fig. 3a–c has been arbitrarily divided into four quarters, defined by PC1 = PC2 = 0.15 a.u., for confining 98% of the control spectra (black dots) into the lower-left quadrant. According to this procedure, three spectral groups have been defined. Spectral group 1 (lower-left quadrant) collects about 70% of the acquired spectra. It contains 98% of the control spectra (black dots) and the large majority of the spectra of animals exposed to AgNCs (cyan dots for AgNCs 3.6 μg Ag per g leaf, 80%, and navy dots for AgNCs 36 μg Ag per g leaf, 86%) and to both sizes of AgNSs at the lower concentrations (light green for AgNS_5–6 0.3 μg Ag per g leaf, 89%, and pink red AgNS_11–12 0.28 μg Ag per g, 76%). Spectral group 2 (upper-left quadrant) is mostly composed of the remaining spectra of animals exposed to AgNCs and to both size of AgNSs at lower concentrations, accounting for about 9% of the acquired dataset. Finally, the spectral group 3 (lower-right quadrant) collects about 20% of the acquired spectra and it contains almost 70% of the spectral dataset collected from glands of animals exposed to AgNS_5–6 3 μg Ag per g leaf (dark grey dots) and about 60% of the spectral dataset collected from the glands of animals exposed to AgNS_11–12 2.8 μg Ag per g leaf (dark red dots). Spectra belonging to the upper-right quadrant are less than 1% of the total dataset, and they will not be considered from here on. The distribution of the spectra of each treatment among the aforementioned spectral groups is summarized in panel d of Fig. 4.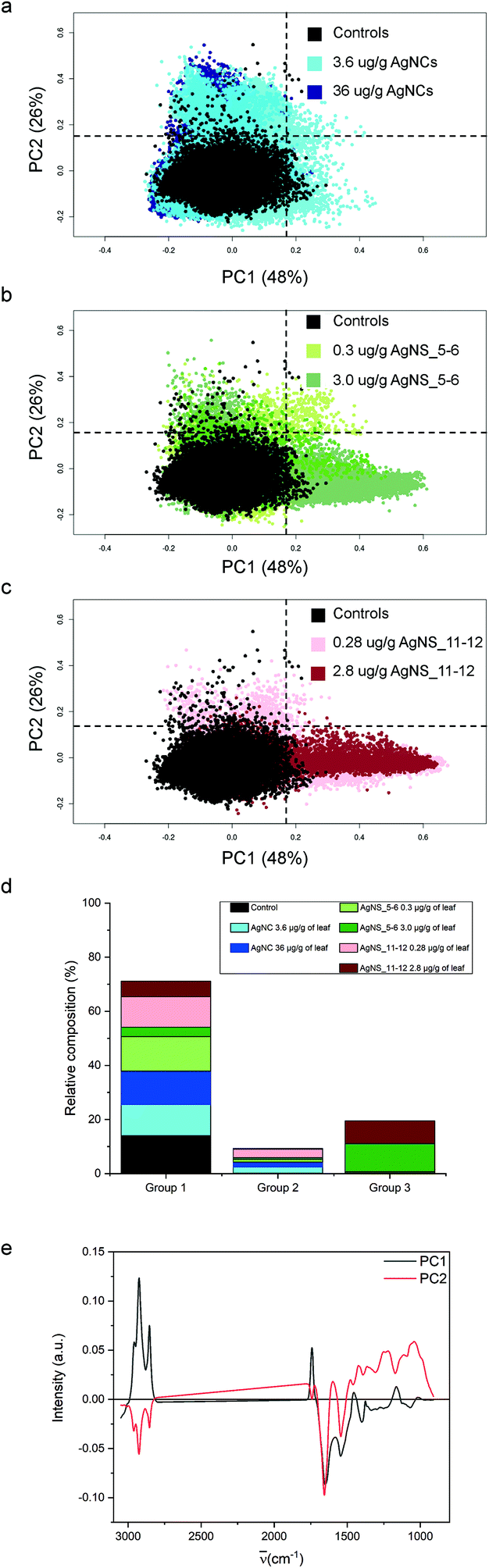 | ||
| Fig. 4 PCA of FTIR imaging data collected from controls and exposed samples. a–c, PC2 vs. PC1 PCA scatter plot of control scores (black) compared to AgNP-exposed tissues: 3.6 Ag NCs per g of leaf (yellow), 36 μg Ag NCs/g of leaf (dark yellow), 0.3 AgNS_5–6 per g of leaf (green), 3.0 AgNS_5–6/g of leaf (dark green), 0.28 AgNS_11–12 AgNS/g of leaf (red), 2.8 AgNS_11–12 AgNS g−1 of leaf (dark red). d, Relative distribution of the seven experimental groups among spectroscopic groups 1, 2 and 3. e, PC1 and PC2 loading plots. | ||
By looking at the spectral features of loading 1 in Fig. 4e (black continuous line), it is possible to deduce that the exposure to the highest concentrations of AgNSs caused an increase in the phospholipid content of the spectral group 3, as shown by the positive signals in the 2800–3000 cm−1 spectral region (asymmetric and symmetric stretching of methyl and methylene moieties) and at 1740 cm−1 (carbonyl ester band of phospholipids).38 The other detectable effect is the decrease in the content of proteins, as seen in the negative signals of both the amide I and amide II bands, associated to the normal modes of vibrations of the peptide backbone. The negative profile cantered at around 1400 cm−1 further confirms the protein trend, as this is the spectral region diagnostic for methylene groups from aminoacids’ lateral chains39 and carbonyl groups of aspartic and glutamic acids.40 It is also possible to see a negative profile of the first loading in the spectral region 1340–1220 cm−1. Since phospholipid content increases in spectral group 3, a reduced protein and nucleic acid content should be considered responsible for this trend. Lastly, the spectral component centered at about 1080 cm−1, assigned to symmetric stretching mode of phosphate moieties of nucleic acids, consistently diminishes. These data are also confirmed by univariate analysis, the results of which are reported in Table S1 of the ESI.†
The spectral analyses of the animals exposed to AgNCs do not show a massive dose-dependent alteration in the chemical features of the cells: only ∼15% of the spectra from each dataset of AgNCs is separated from controls, mainly along PC2, contributing to spectroscopic group 2. In Fig. 4b and c, we can see also that the tissues exposed to a low dose of AgNSs slightly spread along PC2: this represents only 6% of all the measurements on the animals exposed to the lower concentration of AgNS_5–6 and 22% of those exposed to AgNS_11–12 nm. From the analysis of the spectral shape of the loading 2, it is possible to suggest that the exposure to AgNCs and to low concentrations of AgNSs partially affects tissue proteins, as can be seen by the negative profile in the amide I region. Moreover, the sharpness of the negative peak at ∼1654 cm−1, conventionally assigned to alpha-helix proteins, let us postulate the unfolding or misfolding of alpha-helix structures. These treatments also have an effect in the low frequency part of the spectrum. A generalized spectral increase in the 1300–1000 cm−1 region led us to infer increased nucleic acids and/or carbohydrate. Indeed, the positive spectral shape in the wavelength region centered at ∼1050 cm−1, considered diagnostic for carbohydrates, let us assume an increase in carbohydrate metabolism upon NP exposure for the spectral group 2. Present data are confirmed also by univariate analysis (see Table S1 in the ESI†).
In Fig. 5, visible light microscopy images of three representative samples can be compared with the PCA-generated heat images (a–c control, d–f 36 μg AgNC per g of leaf and g–i 2.8 μg AgNS_11–12 per g of leaf). Heat images were obtained by assigning a colour gradient to the PCA scores in such a way that the regions of the tissues that have higher values in one component are red and those that are more similar to the controls are blue. Both PC1 and PC2 images of the control shown in Fig. 5b and c are blue since control animals are not affected by silver exposure. Comparing exposed animals, the PC1 image of the sample fed with 36 μg AgNC per g per leaf is blue, similarly to the controls, while red areas can be mainly seen in the PC2 image. Conversely, the PC2 image of the sample fed with 2.8 μg AgNS_11–12 per g per leaf is blue, while red areas can be mainly seen in the PC1 image. Therefore, the red areas in the PC1 images are the ones more affected by high doses of AgNSs, whereas in PC2 maps they are related to the effects of AgNC exposure, as expected. In addition, the plots could allow the areas of the tissue more or less affected by the AgNP treatment to be identified. AgNP exposure does not appear to affect the entire tissue: there are areas where Ag causes a higher degree of biochemical modification (redder) and areas not affected at all (bluer). Unfortunately, due to the nature of the cryo-slices, the morphology of these relatively thin slices does not allow to identify as B or S cells of the hepatopancreas and therefore the results could not be directly correlated to the PIXE analysis.
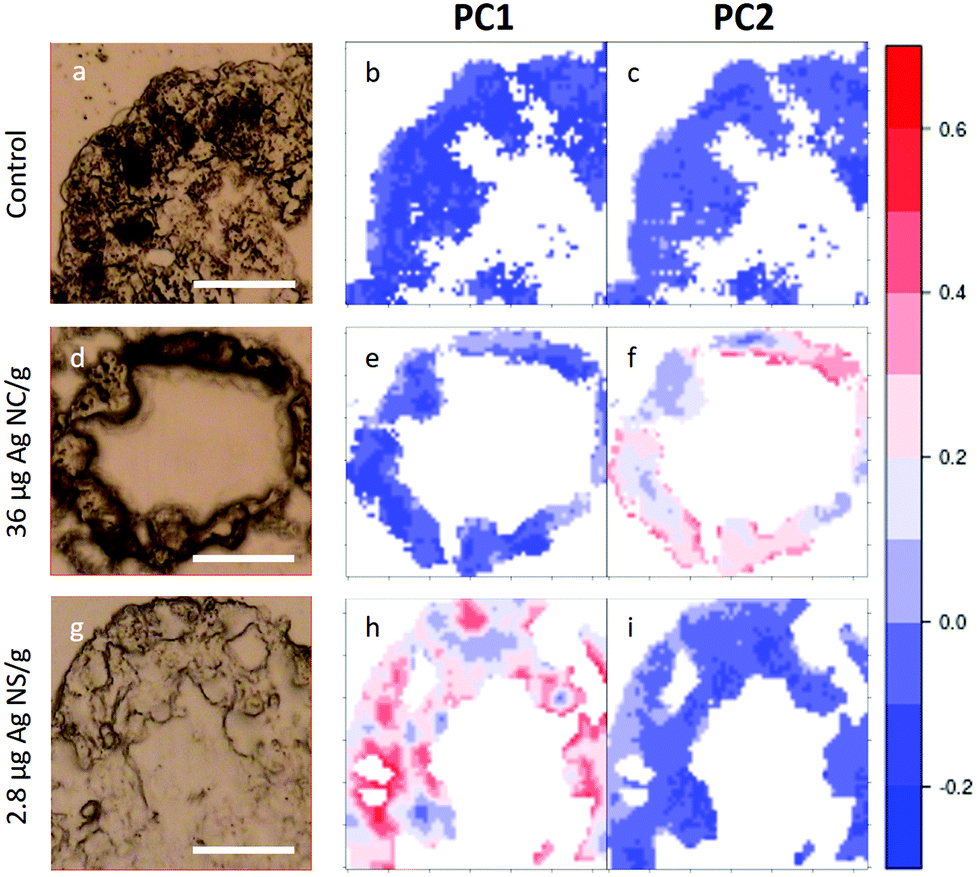 | ||
| Fig. 5 PCA heat maps of controls and exposed samples. a–c, Light microscopy image and PC1 and PC2 images of a representative section of digestive gland of controls; d–f, Light microscopy image and PC1 and PC2 images of a representative section of digestive gland of an animal fed with 36 μg AgNC/g per leaf; g–i, light microscopy image and PC1 and PC2 images of sample a representative section of digestive gland of an animal fed with 2.8 μg AgNS_11–12 per g of leaf. The scale bar, the white line in visible images, is 50 microns. Please see more images of PCA heatmaps in the ESI (Fig. S4†). | ||
However, they do show that the results presented in Fig. 4 are not animal dependent but rather tissue-location driven, and that AgNSs at different concentrations of both sizes affect larger areas of the digestive glands more severely in the animal model P. scaber.
Discussion
Terrestrial isopods (Porcellio scaber, Isopoda, Crustaceae) are suitable test organisms in terrestrial ecotoxicology and ecophysiology, due to their well-known biology and physiology, to the relative ease of laboratory maintenance, and to their important ecological role as decomposers of organic material.41 For the purpose of this work, the animals were fed with AgNCs at two different concentrations and AgNSs of two different sizes and two concentrations. None of the used AgNPs induced toxic effects at the whole organism level: weight, feeding rate and food assimilation efficiency did not change significantly between control and treated groups. However, exposure to both AgNCs and AgNSs resulted in accumulation of Ag in the hepatopancreatic epithelium, following an accumulation trend that primarily dependent on the exposure-dose, as revealed by AAS of digestive glands (see Fig. 2). PIXE analysis also revealed Ag accumulated in the digestive gland of isopods in a dose-dependent manner, co-localizing with Cu in the metal storage granules of S-cells of the digestive gland epithelium (see Fig. 3 and Table 2). It has to be highlighted that there is no literature evidence of NPs directly entering hepatopancreatic cells, as a consequence of cell membrane damage or endocytosis, for both P. scaber42 or other isopods.43 In addition, it was also proven that when fed with NPs, P. scaber intracellularly accumulates amounts of metals which corresponds to the soluble metal fraction.44,45 Similarly, isopods were previously shown to assimilate Ag from sparingly soluble Ag sulfides (Ag2S) that are formed from AgNPs during wastewater-treatment.46 It was therefore concluded that NPs dissolve in the digestive tract of P. scaber, and dissolved ions are taken up by hepatopancreatic cells, that store them in metalliferous granules of S-cells.47,48 Indeed, assimilation of ingested metal ions in S-cells is a well-known metal detoxification mechanism in isopods, that has been specifically proven by our group for AgNPs by combining PIXE and TEM analysis on digestive gland cross sections.49 Therefore, it is possible to claim that the silver distribution imaged by PIXE and AAS data from digestive glands mostly represent the dissolved silver ions and their cellular uptake.However, AgNP dissolution does not fully justify ASS and PIXE data. As a matter of fact, AgNSs with different sizes have comparable dissolution percentages in vitro (see Table 1), but Ag ions from smaller AgNPs accumulate more under in vivo conditions (see Fig. 2 and Table 2). Therefore, a dependence of the Ag accumulation from AgNP shape and size can be postulated.
Taking into consideration the biochemical effects induced by the exposure to sub-toxic concentrations of AgNPs as revealed by FTIR imaging, the explanation of AgNP interactions with cells becomes even more complex. The six different AgNP exposure groups resulted in two distinct types of FTIR molecular fingerprints with respect to spectroscopic group 1, associated with a NP response that cannot be fully explained by the uptake of silver ions in the hepatopancreatic cells. The first response type, associated to spectral group 2, was found as a result of exposure to AgNCs at both concentrations and exposure to AgNSs at the lower concentrations. The second type, associated to spectral group 3, was found in the tissue of animals exposed to both (smaller and larger) AgNSs at the higher exposure concentration. As a matter of fact, if we compare the effects caused by AgNPs in the same Ag concentration range (the lower concentration of AgNCs and the higher exposure concentration of both sizes of AgNSs), different molecular fingerprints have been highlighted by PCA analysis (see Fig. 4). As the dissolution of silver is the major player in inducing sub-cytotoxic effects, we would expect the aforementioned NPs to belong to the same spectral group, but this is not the case. Furthermore, in the tissue the highest accumulation of Ag ions was observed for AgNC 36 μg Ag per g leaf, but only a small portion of the samples (about 15%) were affected by the exposure to an extent and manner comparable with the lower AgNC dose. PCA images in Fig. 5 also confirm that a low percentage of the tissue was actually affected by similar molecular changes for animals fed with AgNCs 36 μg/g leaf, and to a lesser extent with respect to animals fed with AgNSs 2.8 μg/g leaf. Consequently, the in vitro dissolution potential solely does not explain the observed in vivo effects.
Indeed, size and shape of the AgNPs seem also to play a role in defining the pattern of biochemical modifications associated to NP exposure. Cubic AgNPs that exert sub-cytotoxic effects belong to spectroscopic group 2, while Ag nanospheres of both sizes concentrate into spectroscopic group 3. However, the size also plays a role, being a relevant fraction of the larger AgNSs dosed at 0.28 μg/g of leaf part of spectroscopic group 2. However, the size of these nanospheres, ranging from 11 to 12 nm in diameter, is smaller than that of NCs and their in vivo accumulation is lower than AgNCs and AgNS_5–6 and comparable to that of AgNS_11–12 dosed 2.8 μg/g of leaf.
Consequently, neither the shape nor the size solely explains the observed effects.
A number of studies have reported that either (i) dissolution is the main driver of toxicity6–8 or (ii) size matters,6,14,18 while others (iii) relate the effects predominately to the shape.20,21,23 Indeed, the FTIRI results here presented confirm that the intrinsic properties of silver nanoparticles, i.e. size, shape, concentration and release of ions from dissolved nanoparticles, jointly contribute to the molecular alterations of the digestive gland tissue of isopods (Porcellio scaber, Isopoda, Crustaceae) after oral exposure to subtoxic levels, and that Ag accumulation in tissues is just one of the parameters not directly related to the extent of the changes at a molecular level.
Analysing the specific spectral features characterizing the spectral groups 2 it is possible to recognize the unfolding or misfolding of alpha-helix proteins and an increase in both nucleic acids and carbohydrates, while for the spectral group 3, the sub-toxic effects can be interpreted as an accumulation of phospholipids and a down regulation of protein synthesis. Many other authors have also used the FTIR method to show that different chemicals are affecting major biochemical constituents,50 but FTIR tissue analyses from in vivo studies with nanoparticles are scarce. In our previously published studies,27,28 other nanomaterials provoked a different pattern of molecular response in the same experimental set-up (a 14-day feeding exposure of terrestrial isopods). Novak et al.28 studied the effects of WOx nanowires (nano-WOx) and Romih et al.34 studied the effects of ZnO2 NPs and ZnCl2 salt. When the molecular response of spectroscopic group 2 as described in this work is compared with the results of these two studies, Novak et al.28 did not find any effects on proteins or any increase of carbohydrates. Romih et al.34 reported an altered conformation of proteins, mostly random domains and altered alpha-helix folding patterns. However, they did not find alterations in the intensity of both Amide I and Amide II as is the case in our AgNP study. In addition, Romih et al.34 described increased carbohydrates, but only in the case of ZnCl2 exposure. In general, more pronounced spectra alterations were observed after ZnCl2 ingestion, letting us conclude that Zn ions are the main cause of the observed effects. When the results of Novak et al.51 and Romih et al.34 are compared to the molecular pattern of spectroscopic group 3 as shown in the present study, all three studies reported alterations in the signal at 1740 cm−1 annotating more phospholipids. Changes in the amount of phospholipids are an indication of changes in membrane fluidity, effects which have been caused by different stressors as already measured by FTIR in other studies.52–55 A similar effect on lipids was observed in the study by Vileno et al.,56 where FTIR was used to study the effects of fullerols on cells. Those authors monitored oxidative stress in cells induced via fullerols under visible light illumination and observed an increase in the bands linked with lipid peroxidation and protein phosphorylation.
The survey conducted in our study highlights that it is probably impossible to find standardized sub-toxicity markers that could be applied to any sample and to any NP, while it is more reliable to admit that the response is both animal and NP specific. This specificity can be clearly highlighted and partially disentangled by FTIR imaging, which can also provide spatially-resolved information, to be correlated to other microscopy techniques.
Conclusions
The present paper supports the evidence that AgNP effects at the sub-toxic concentration are the result of the interplay between size, shape and dissolution of ions from NPs and their combined interactions with tissue. The message of this work, and other similar studies, is that we should stop looking for a single NP characteristic responsible for biological effects; instead, we must search for methods and approaches able to reveal the biological consequences of the combinatorial effects of different NP characteristics that would allow us to discriminate low hazard NPs from highly biologically toxic NPs. FTIR imaging seems a good candidate in this respect.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge Elettra Sicnrotrone Trieste for provision of synchrotron radiation facilities (proposal N. 20115304). The study was partly funded by NanoFASE (EC H2020 grant agreement No 646002).Notes and references
- M. E. Vance, T. Kuiken, E. P. Vejerano, S. P. McGinnis, M. F. Hochella, D. Rejeski and M. S. Hull, Beilstein J. Nanotechnol., 2015, 6, 1769–1780 CrossRef CAS PubMed.
- R. D. Handy, G. Cornelis, T. Fernandes, O. Tsyusko, A. Decho, T. Sabo-Attwood, C. Metcalfe, J. A. Steevens, S. J. Klaine, A. A. Koelmans and N. Horne, Environ. Toxicol. Chem., 2012, 31, 15–31 CrossRef CAS PubMed.
- Y.-J. Lee, J. Kim, J. Oh, S. Bae, S. Lee, I. S. Hong and S.-H. Kim, Environ. Toxicol. Chem., 2012, 31, 155–159 CrossRef CAS PubMed.
- C. Levard, E. M. Hotze, G. V. Lowry and G. E. Brown, Environ. Sci. Technol., 2012, 46, 6900–6914 CrossRef CAS PubMed.
- F. Gottschalk, T. Sonderer, R. W. Scholz and B. Nowack, Environ. Sci. Technol., 2009, 43, 9216–9222 CrossRef CAS PubMed.
- R. Ma, C. Levard, S. M. Marinakos, Y. Cheng, J. Liu, F. M. Michel, G. E. Brown and G. V. Lowry, Environ. Sci. Technol., 2012, 46, 752–759 CrossRef CAS PubMed.
- Z. Xiu, Q. Zhang, H. L. Puppala, V. L. Colvin and P. J. J. Alvarez, Nano Lett., 2012, 12, 4271–4275 CrossRef CAS PubMed.
- X. Yang, A. P. Gondikas, S. M. Marinakos, M. Auffan, J. Liu, H. Hsu-Kim and J. N. Meyer, Environ. Sci. Technol., 2012, 46, 1119–1127 CrossRef CAS PubMed.
- A. Ivask, I. Kurvet, K. Kasemets, I. Blinova, V. Aruoja, S. Suppi, H. Vija, A. Käkinen, T. Titma, M. Heinlaan, M. Visnapuu, D. Koller, V. Kisand and A. Kahru, PLoS One, 2014, 9, e102108 CrossRef PubMed.
- D. E. Gorka, J. S. Osterberg, C. A. Gwin, B. P. Colman, J. N. Meyer, E. S. Bernhardt, C. K. Gunsch, R. T. DiGulio and J. Liu, Environ. Sci. Technol., 2015, 49, 10093–10098 CrossRef CAS PubMed.
- Y. Xiong, M. Brunson, J. Huh, A. Huang, A. Coster, K. Wendt, J. Fay and D. Qin, Small, 2013, 9, 2628–2638 CrossRef CAS PubMed.
- R. Podila and J. M. Brown, J. Biochem. Mol. Toxicol., 2013, 27, 50–55 CrossRef CAS PubMed.
- M. Zhu, G. Nie, H. Meng, T. Xia, A. Nel and Y. Zhao, Acc. Chem. Res., 2013, 46, 622–631 CrossRef CAS PubMed.
- M. Almansour, Q. Jarrar, A. Battah and B. Jarrar, Int. J. Morphol., 2015, 33, 544 CrossRef.
- D. A. Cowart, S. M. Guida, S. I. Shah and A. G. Marsh, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2011, 46, 1122–1128 CrossRef CAS PubMed.
- O. Choi and Z. Hu, Environ. Sci. Technol., 2008, 42, 4583–4588 CrossRef CAS PubMed.
- H.-G. Park and M.-K. Yeo, Mol. Cell. Toxicol., 2014, 10, 401–409 CrossRef CAS.
- T.-H. Kim, M. Kim, H.-S. Park, U. S. Shin, M.-S. Gong and H.-W. Kim, J. Biomed. Mater. Res., Part A, 2012, 100, 1033–1043 CrossRef PubMed.
- K. S. Butler, D. J. Peeler, B. J. Casey, B. J. Dair and R. K. Elespuru, Mutagenesis, 2015, 30, 577–591 CrossRef CAS PubMed.
- S. Pal, Y. K. Tak and J. M. Song, Appl. Environ. Microbiol., 2007, 73, 1712–1720 CrossRef CAS PubMed.
- M. Visnapuu, U. Joost, K. Juganson, K. Künnis-Beres, A. Kahru, V. Kisand and A. Ivask, BioMed Res. Int., 2013, 2013, 819252 Search PubMed.
- Y. Syu, J.-H. Hung, J.-C. Chen and H. Chuang, Plant Physiol. Biochem., 2014, 83, 57–64 CrossRef CAS PubMed.
- S. George, S. Lin, Z. Ji, C. R. Thomas, L. Li, M. Mecklenburg, H. Meng, X. Wang, H. Zhang, T. Xia, J. N. Hohman, S. Lin, J. I. Zink, P. S. Weiss and A. E. Nel, ACS Nano, 2012, 6, 3745–3759 CrossRef CAS PubMed.
- S. Komarneni, D. Li, B. Newalkar, H. Katsuki and A. S. Bhalla, Langmuir, 2002, 18, 5959–5962 CrossRef CAS.
- A. R. Siekkinen, J. M. McLellan, J. Chen and Y. Xia, Chem. Phys. Lett., 2006, 432, 491–496 CrossRef CAS PubMed.
- J. Vidmar, T. Buerki-Thurnherr and K. Loeschner, J. Anal. At. Spectrom., 2018, 33, 752–761 RSC.
- T. Romih, A. Jemec, S. Novak, L. Vaccari, P. Ferraris, M. Šimon, M. Kos, R. Susič, K. Kogej, J. Zupanc and D. Drobne, Nanotoxicology, 2015, 1–9 Search PubMed.
- S. Novak, D. Drobne, L. Vaccari, M. Kiskinova, P. Ferraris, G. Birarda, M. Remškar and M. Hočevar, Environ. Sci. Technol., 2013, 47, 11284–11292 CrossRef CAS PubMed.
- S. Sorieul, P. Alfaurt, L. Daudin, L. Serani and P. Moretto, Nucl. Instrum. Methods Phys. Res., Sect. B, 2014, 332, 68–73 CrossRef CAS.
- S. Lupi, A. Nucara, A. Perucchi, P. Calvani, M. Ortolani, L. Quaroni and M. Kiskinova, J. Opt. Soc. Am. B, 2007, 24, 959–964 CrossRef CAS.
- C. Beleites and V. Sergo, HyperSpec Package Handle Hyperspectral Data Sets R R Package Version 0 98–201209223httphyperspecr-Forg.-Proj., 2013.
- F. Murtagh, Comput. J., 1983, 26, 354–359 CrossRef.
- R. Sibson, Comput. J., 1973, 16, 30–34 CrossRef.
- T. Romih, A. Jemec, S. Novak, L. Vaccari, P. Ferraris, M. Šimon, M. Kos, R. Susič, K. Kogej, J. Zupanc and D. Drobne, Nanotoxicology, 2016, 10, 462–470 CrossRef CAS PubMed.
- P. Billaud, J.-R. Huntzinger, E. Cottancin, J. Lermé, M. Pellarin, L. Arnaud, M. Broyer, N. D. Fatti and F. Vallée, Eur. Phys. J. D, 2007, 43, 271–274 CrossRef CAS.
- T. Romih, S. B. Hočevar, A. Jemec and D. Drobne, Electrochim. Acta, 2016, 188, 393–397 CrossRef CAS.
- S. P. Hopkin and M. H. Martin, Tissue Cell, 1982, 14, 703–715 CrossRef CAS PubMed.
- Z. Movasaghi, S. Rehman and D. I. ur Rehman, Appl. Spectrosc. Rev., 2008, 43, 134–179 CrossRef CAS.
- N. Fujioka, Y. Morimoto, T. Arai and M. Kikuchi, Cancer Detect. Prev., 2004, 28, 32–36 CrossRef PubMed.
- H. Fabian, M. Jackson, L. Murphy, P. H. Watson, I. Fichtner and H. H. Mantsch, Biospectroscopy, 1995, 1, 37–45 CrossRef CAS.
- C. A. M. van Gestel, ZooKeys, 2012, 176, 275–296 CrossRef PubMed.
- S. Novak, D. Drobne, M. Golobič, J. Zupanc, T. Romih, A. Gianoncelli, M. Kiskinova, B. Kaulich, P. Pelicon, P. Vavpetič, L. Jeromel, N. Ogrinc and D. Makovec, Environ. Sci. Technol., 2013, 47, 5400–5408 CrossRef CAS PubMed.
- P. S. Tourinho, C. A. M. van Gestel, A. J. Morgan, P. Kille, C. Svendsen, K. Jurkschat, J. F. W. Mosselmans, A. M. Soares and S. Loureiro, Ecotoxicology, 2016, 25, 267–278 CrossRef CAS PubMed.
- M. Golobič, A. Jemec, D. Drobne, T. Romih, K. Kasemets and A. Kahru, Environ. Sci. Technol., 2012, 46, 12112–12119 CrossRef PubMed.
- Ž. Pipan-Tkalec, D. Drobne, A. Jemec, T. Romih, P. Zidar and M. Bele, Toxicology, 2010, 269, 198–203 CrossRef PubMed.
- S. Kampe, R. Kaegi, K. Schlich, C. Wasmuth, H. Hollert and C. Schlechtriem, Environ. Toxicol. Chem., 2018, 37, 1606–1613 CrossRef CAS PubMed.
- T. Romih, B. Drašler, A. Jemec, D. Drobne, S. Novak, M. Golobič, D. Makovec, R. Susič and K. Kogej, Sci. Total Environ., 2015, 508, 76–84 CrossRef CAS PubMed.
- T. Romih, A. Jemec, M. Kos, S. B. Hočevar, S. Kralj, D. Makovec and D. Drobne, Environ. Pollut., 2016, 218, 957–964 CrossRef CAS PubMed.
- Ž. P. Tkalec, D. Drobne, K. Vogel-Mikuš, P. Pongrac, M. Regvar, J. Štrus, P. Pelicon, P. Vavpetič, N. Grlj and M. Remškar, Nucl. Instrum. Methods Phys. Res., Sect. B, 2011, 269, 2286–2291 CrossRef.
- K. le Roux, L. C. Prinsloo and D. Meyer, Spectrochim. Acta, Part A, 2015, 138, 321–330 CrossRef CAS PubMed.
- S. Novak, D. Drobne, L. Vaccari, M. Kiskinova, P. Ferraris, G. Birarda, M. Remškar and M. Hočevar, Environ. Sci. Technol., 2013, 47, 11284–11292 CrossRef CAS PubMed.
- G. Birarda, D. E. Bedolla, E. Mitri, S. Pacor, G. Grenci and L. Vaccari, Analyst, 2014, 139, 3097–3106 RSC.
- P. Demir, S. B. Akkas, M. Severcan, F. Zorlu and F. Severcan, Appl. Spectrosc., 2015, 69, 154–164 CrossRef CAS PubMed.
- S. Mahajan and N. Tuteja, Arch. Biochem. Biophys., 2005, 444, 139–158 CrossRef CAS PubMed.
- K. Rajapakse, D. Drobne, J. Valant, M. Vodovnik, A. Levart and R. Marinsek-Logar, J. Hazard. Mater., 2012, 221–222, 199–205 CrossRef CAS PubMed.
- B. Vileno, S. Jeney, A. Sienkiewicz, P. R. Marcoux, L. M. Miller and L. Forró, Biophys. Chem., 2010, 152, 164–169 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8an01387j |
| ‡ Current address: SEYENS Ltd., Ljubljana, Slovenia. |
| § Current address: Adolphe Merkle Institute, University of Fribourg, Fribourg, Switzerland. |
| ¶ Current address: NRE Research s.r.l., AREA Science Park, Basovizza, Trieste, Italy. |
| This journal is © The Royal Society of Chemistry 2019 |