
“Fix and assay”: separating in-cellulo sphingolipid reactions from analytical assay in time and space using an aldehyde-based fixative†
 a
and
Nancy L.
Allbritton
*ab
a
and
Nancy L.
Allbritton
*ab
Abstract
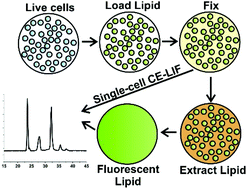
Chemical cytometry using capillary electrophoresis (CE) is a powerful tool for measuring single-cell enzyme activity. However, these measurements are often confounding as dynamic processes within cells rapidly change depending on environment, meaning that cell handling, transport, and storage can affect signaling pathways and alter results. To meet these challenges, we describe a method utilizing aldehyde fixation to simultaneously terminate cellular reactions across a population, freezing reaction results in time prior to analytical analysis. Fluorescent sphingosine was loaded into cells of different lineages (leukemia and lymphoma cell lines and primary leukemia cells) and allowed to react before fixing. The remaining sphingosine and any products formed were then quantified with chemical cytometry utilizing CE. When cells were loaded with sphingosine followed by glyoxal fixation and immediate analysis, 55 ± 5% of lipid was recoverable compared to an unfixed control. Storage of fixed cells for 24 h showed no statistical differences in total amount of recoverable sphingolipid compared to samples analyzed immediately after fixation—though there was a difference in recovery of low-abundance products. Sphingosine kinase activity decreased in response to inhibitor treatment compared to treatment with a DMSO vehicle (21 ± 3% product formed in inhibitor-treated cells vs. 57 ± 2% in control cells), which was mirrored in single-cell measurements. This “fix and assay” strategy enables measurement of sphingosine kinase activity in single cells followed by subsequent analytical assay separated in space and time from reaction initiation, enabling greater temporal control over intracellular reactions and improving future compatibility with clinical workflow.
- This article is part of the themed collection: Next wave advances in single cell analyses
 Please wait while we load your content...
Please wait while we load your content...

