
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn-line whole blood fractionation for Raman analysis of blood plasma
Moritz
Matthiae
 ,
Xiaolong
Zhu
,
Rodolphe
Marie
and
Anders
Kristensen
*
,
Xiaolong
Zhu
,
Rodolphe
Marie
and
Anders
Kristensen
*
Department of Micro- and Nanotechnology, Technical University of Denmark, 2800 Kongens-Lyngby, Denmark. E-mail: akri@dtu.dk
First published on 16th November 2018
Abstract
Blood plasma evaluation has high significance in clinical diagnostics. Current schemes involve the preparation of blood plasma by centrifugation of whole blood followed by electrochemical or spectroscopic analysis. However, centrifugation is often too time-consuming for application in clinical emergency and point-of-care settings. We propose to combine microfluidic, instantaneous plasma fractionation with localized spectroscopic methods for in-line analysis. As an example, we present confocal Raman spectroscopy in fractionated plasma domains at two different Raman excitation wavelengths. Resonance Raman spectroscopy with laser excitation at 408 nm allows the specific detection of free hemoglobin in blood plasma at concentrations above 22 mg dl−1 (level of detection). Consequently, we are able to accurately resolve the range of clinical relevance regarding hemolysis. At near-infrared excitation (785 nm) we furthermore demonstrate the acquisition of characteristic Raman spectra of fractionated blood plasma in the microfluidic setting. These spectra can serve as starting point for a multi-parameter regression analysis to quantify a set of blood plasma parameters from a single Raman spectrum. The combined microfluidics and Raman spectroscopy method is non-destructive and has a whole blood consumption of less than 100 μl per hour. It thus allows for continuous in-line blood plasma monitoring.
1 Introduction
Exact and reliable blood assay is key in clinical diagnostics. Every day millions of blood samples are drawn from patients around the globe to provide physicians with crucial information on the patients’ health status. Point-of-care blood testing, in particular in emergency settings, requires particularly fast sample throughput and analysis, on a timescale of less than one minute. In this study we demonstrate an in-line blood plasma analysis, where we employ optical sensing inside a locally expanded plasma domain in microfluidic whole blood flow. With this method, time-consuming centrifugation of whole blood is circumvented. Furthermore, continuous blood sample consumption can be kept below 100 μl per hour due to small microfluidic channel dimensions, thereby enabling continuous plasma monitoring or analysis in a prick test. Fig. 1 illustrates the non-permanent, yet instantaneous cell-free plasma domain creation alongside the boundaries of the microfluidic channel expansion. Confocal laser illumination on streaming red blood cells and on fractionated plasma results in distinct Raman spectra.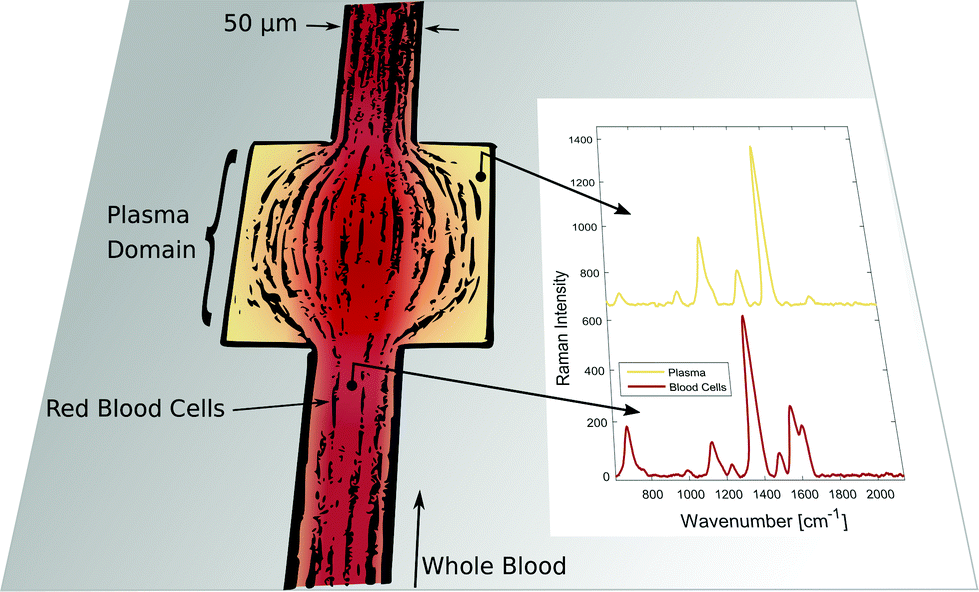 | ||
| Fig. 1 Instantaneous blood plasma fractionation appears adjacent to the channel walls of a geometric expansion in microfluidic whole blood flow. Local Raman spectroscopy on plasma domains (orange) is different compared to domains of streaming whole blood (red) where Raman scattering originates from cellular hemoglobin.4 The Raman excitation wavelength is 408 nm. | ||
Blood flow in micro-capillaries and microchannels has been the subject of research for many years as it provides information on in vivo blood flow in microcirculation.1,2 Whole blood can be considered as a suspension of erythrocytes in a Newtonian fluid,3 namely blood plasma. Due to the velocity gradient across a shear flow in a microchannel and the consequent hydrodynamical lift, deformable cells tend to migrate away from the channel boundaries towards the middle of the channel resulting in cell-free plasma layers adjacent to the boundaries. Inside the microchannels of 10–300 μm in diameter, blood cells literally float on the plasma layer keeping the blood cells at a distance of about 1.5 μm from the walls.1,3 This particularity implies the reduction of apparent dynamic blood viscosity in the microchannels of the stated diameter (Fahraeus–Lindqvist effect). The extent of the plasma layer can be manipulated by local constriction or expansion of the microfluidic blood channel.5 Extended plasma domains arise in geometric singularities of the microfluidic channel, such as cavities or local expansions. These principles have been combined with various microfluidic approaches for passive, permanent plasma extraction from diluted6,7 and undiluted8 whole blood. Here we focus on local channel expansions for non-permanent blood plasma domain creation. These domains must be large enough to enable in-line spectroscopic probing.
Confocal Raman microscopy enables localized chemical sensing in the expanded cell-free plasma region. In general, the combination of microfluidics and Raman spectroscopy has a large number of applications as reviewed by Chrimes et al.,9 for example monitoring of chemical reactions in microfluidic reactors,10 waveguide confined integration of Raman spectroscopy on microfluidic chips11 and surface-enhanced Raman techniques in microfluidics.12,13 Specifically, Raman analysis of blood and blood components has shown potential for various medical applications as summarized in a recent comprehensive review article.14 In particular, quantitative multi-component Raman analysis of blood constituents like glucose, cholesterol, triglyceride, urea, total protein and albumin was successfully performed on liquid blood plasma15 and whole blood16 by means of Partial Least Squares (PLS) regression. The starting point for these regression studies are background-corrected Raman spectra from plasma or serum excited at a near-infrared wavelength and acquired in the spectral window from 600 cm−1 to 1800 cm−1.
Classification studies of blood plasma Raman experiments show encouraging results too. For example in cancer diagnostics, it is possible to discriminate different cancer types17–19 from a respective healthy reference group as well as different tumor phases.20 Blood plasma or serum is preferred against whole blood in most experiments in order to avoid interference due to red blood cells.21 In order to enhance the inherently weak Raman scattering intensities, surface-enhanced Raman spectroscopic (SERS) substrates are employed throughout these studies, thereby generating very clear Raman spectra with an enhanced signal-to-noise ratio at reduced acquisition times.17,20 Liquid mixtures of whole blood or blood plasma with SERS colloidal nanoparticles22 are often used. They are compatible with the microfluidic Raman setup of this paper.
As a direct proof of plasma Raman response, we focus on a critical clinical parameter which can be determined from blood plasma only: the concentration of free hemoglobin in whole blood. A concentration of about 100 mg dl−1 free hemoglobin in blood plasma is considered as the clinical limit above which unnaturally intensified rupture of erythrocytes (hemolysis) undoubtedly occurs.23 We demonstrate specific hemoglobin sensing in fractionated blood plasma by Raman spectroscopy and resolve the concentration range of clinical relevance for hemolysis. This sensitivity can only be reached because hemoglobin is a strong Raman scatterer if the excitation laser wavelength is chosen spectrally close to the Soret band absorption of the molecule to exploit the resonance Raman effect.24 The oscillating mode of the central porphyrin ring structure of hemoglobin has a large Raman cross-section resulting in a dominant peak in the Raman spectrum at about 1375 cm−1.4,25 In recent years hemoglobin Raman research included for example single molecule detection of hemoglobin by SERS,26 transdermal oxygen saturation measurements of hemoglobin,27 multi-dimensional signatures in forensic identifications28 and hemoglobin fingerprints in whole blood samples.29 The different oxidation states of oxyhemoglobin, deoxyhemoglobin and met-hemoglobin are distinguishable by their Raman spectra.30,31 Because blood samples have been exposed to atmospheric oxygen partial pressure, we focus on oxyhemoglobin in our hemolysis study.
2 Experimental
Blood samples
We receive bovine whole blood samples from a company (SSI Diagnostica A/S, Hillerod, Denmark). All animal procedures were performed in accordance with the EU-directive 2010/63/EU on the protection of animals used for scientific purposes as well as the Danish order regarding animal experimentation, and approved by the Danish Veterinary and Food Administration. We store the blood at 4 °C until use within two days from extraction. A state-of-the-art blood gas analyzer (ABL90, Radiometer Medical ApS, Copenhagen, Denmark) is used to determine the absolute hemoglobin concentration of this blood as well as its hematocrit. Commonly measured hemoglobin concentrations are in the range 12–16 g dl−1. In a freezer, we fully hemolyze a small fraction of this blood sample so that it can serve as free hemoglobin donor. We then spike non-hemolyzed whole blood with a known amount of free hemoglobin in order to mimic hemolysis. While considering the respective hematocrit value we prepare whole blood samples with free hemoglobin in the virtual plasma phase at concentrations ranging from 0 mg dl−1 to 400 mg dl−1. Furthermore, bovine plasma is generated by centrifugation of the initial whole blood at 5000 rpm for 10 minutes. We prepare plasma samples spiked with free hemoglobin in the same range of concentrations.Microfluidic channels
We prepare rectangular microfluidic channels of 50 μm in width, 9 mm in length and either 20 μm or 40 μm in height in polydimethylsiloxane (PDMS). Initially, a silicon wafer is manufactured by UV-lithography (SUESS MicroTEC, Garching, Germany) and deep reactive ion etching (DRIE Pegasus, SPTS, Allentown, United States). This wafer is a negative copy of the final microfluidic channel. Subsequently, an anti-stiction coating of fluorocarbon is deposited onto the structured silicon surface in a molecular vapor deposition process (MVD 100, Applied Microstructures Inc., Newport, South Wales). Liquid PDMS – homogeneously mixed with ten weight percent of the corresponding developer – is cast onto the silicon stamp and left in an oven at 75 °C for four hours. The fluidic inlet and outlet ports are introduced into the hardened PDMS chip which is then bonded to a fused silica cover slice of 200 μm in thickness. By using fused silica instead of glass, we avoid characteristic Raman emission from the window material, in particular at 785 nm excitation.32 Oxygen plasma is applied for the oxidative activation of the surfaces33 enabling a stable bond and thus a consistent microfluidic chip. The microfluidic channel is prepared for blood flow experiments by wetting it with buffer solution that matches the blood pH. This buffer is also used as a rinse for channel cleaning in between different whole blood samples. We insert a sample volume of about 30 μl into the inlet reservoir and apply an underpressure in the range of 1–100 mbar from a low-cost vacuum pump to the outlet reservoir in order to make the blood flow. Typical flow rates are 10–100 μl per hour. By using underpressure to drive the flow we make sure that the inlet port is always accessible for fluidic sample exchange and fluidic connectors do not need to be moved. In this way the chip position and hereby the optical alignment is conserved during the sample exchange and consecutive measurements.Microfluidic Raman setup
The microfluidic PDMS chip is mounted in an inverted optical microscope (Nikon-Ti, Shinagawa, Japan), being equipped with a 60× (20×) air objective which has a numerical aperture of 0.85 (0.50) and a working distance of 300 μm (3 mm). Illumination with white light is applied from the top and an EMCCD camera (Evolve 512, Tucson Arizona, United States) captures the movement of the blood stream in microfluidic channels at a rate of 100 frames per second. For Raman excitation, the collimated beam from a 25 mW diode laser (DL405-25, CrystaLaser, Reno Nevada, United States) at the wavelength of 408 nm is coupled to the microscope and focused by the objective to a spot of about 1 μm in the field of view. Alternatively, a 785 nm laser of narrow linewidth (ND08-01 785 nm, Cobolt Lasers, Stockholm, Sweden) can be used for near-infrared Raman experiments. The collimation of the initial laser beam and thereby its focus in the imaged plane can be tuned with a telescope before coupling to the microscope as illustrated in Fig. 2(c). | ||
| Fig. 2 Microfluidic Raman setup. (a) The localized cell-free plasma domain in microfluidic whole blood stream is probed by confocal Raman spectroscopy. (b) The Raman laser penetrates both the microfluidic channel and the PDMS host material. (c) The microfluidic chip is mounted on the translation stage of an inverted microscope with confocal laser excitation at 408 nm. Raman emission is analyzed by an imaging dispersive spectrometer. | ||
The microfluidic flow inside the channel and the laser spot for local Raman excitation are imaged simultaneously by using a Shamrock 303i imaging spectrometer which is equipped with a Newton 920 deep-cooled back-illuminated CCD camera (Andor Technology, Belfast, Northern Ireland). The position of the focused laser spot in the field of view is adjusted such that the locally excited scattering from the sample enters the spectrometer through the insertable 100 μm entrance slit. An appropriate Notch filter (NF405-13 or NF785-33, Thorlabs) suppresses Rayleigh scattered light, such that only the much weaker Raman scattered contribution enters the spectrometer. Spectral dispersion by a grating with 1200 lines per mm (408 nm laser) or 600 lines per mm (785 nm laser) causes a horizontal distribution of detected Raman intensity on the CCD camera. However, different vertical positions on the CCD camera correspond to spatially-offset positions of Raman scattering relative to the position of laser excitation. Spatially-offset intensity reports Raman scattering from a sample volume that is not in focus with the imaged plane of the microscope.34,35
In the case of very low light levels vertical binning of camera pixel intensities allows sufficient detection of Raman photons. Furthermore, we employ moving average smoothing to the acquired Raman spectra and average counts from five horizontally adjacent CCD pixels (adjacent wavelengths), respectively.
Microfluidic modeling
Fig. 3 shows that the patterns of microfluidic blood flow and plasma recirculation depend on the expansion geometry. In order to derive design criteria for the expansion geometry, we perform quasi-3D COMSOL simulations36 of stationary flow in microfluidic channels by applying a two-dimensional creeping flow model and the shallow channel approximation. The modeling of non-Newtonian fluids like whole blood is an advanced task, particularly due to the deformability of red blood cells, their non-spherical shape and their dynamic interactions.3 However, we are interested in the flow properties of the plasma domain which is an incompressible Newtonian fluid. Nevertheless, the flow in the adjacent cell-enriched domain needs to be considered as well. In order to derive approximate boundary conditions for the cell-depleted plasma domain of our interest, we also model the cell-enriched domain as an incompressible Newtonian fluid having identical properties to the plasma. The Reynolds number of our system is low (Re ≈ 0.1) so that inertial terms can be neglected and the 2D Stokes equation in the shallow channel approximation36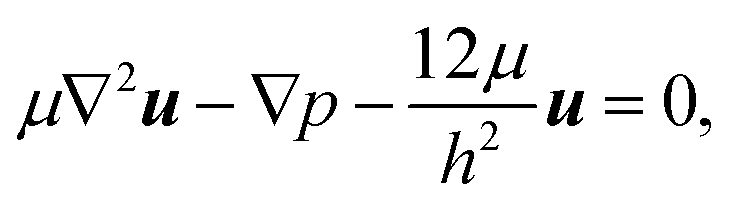 | (1) |
| ∇·u = 0 | (2) |
 | ||
| Fig. 3 Experimental microfluidic whole blood stream in 40 μm deep PDMS channels and localized cell-free domain creation for two different expansion geometries. From left to right, respectively: 10 ms snapshot (a–b), time average of blood flow time series (c–d), standard deviation of blood flow time series (e–f) and creeping flow streamline simulation (g–h). The yellow streamlines indicate the extent of the plasma domain that is fed by the 1.5 μm wide plasma layer of the inlet whole blood flow. | ||
3 Results and discussion
Microfluidic instantaneous plasma domain creation
In a straight microfluidic channel, the plasma layer in the streaming blood is about 1.5 μm wide.1,3 This is too small for confocal probing under moderate numerical apertures. Therefore we aim to locally expand this cell-free layer into extended plasma domains of at least 10 μm in diameter which can be achieved by introducing a local expansion in the otherwise w = 50 μm wide microfluidic channel. Fig. 3 illustrates that the patterns of blood flow and the extent of fractionated plasma domains in the expansion are determined by its geometry. We modify the length L of the expansion while expansion ratio W/w = 3 and channel height h = 40 μm are kept constant. For L = 2w streamlines are continuous and sufficiently expanded plasma domains reach the corners of the expansion. In the case of L = w (lower row in Fig. 3), eddies occur – as analytically described by Moffat37 – which implies that the incoming plasma layer is not largely expanded in the corners of the expansion geometry. Nevertheless, the plasma layer feeds the eddy by diffusion across their common boundary. But due to the small width of the plasma layer inside the expansion, some cells enter the eddy and circulate. This effect becomes most obvious in standard deviation images of the blood flow time series.Confocal laser illumination in the region of a microfluidic eddy is critical. Although this region contains blood plasma, some cells circulate and can get trapped and destroyed by the focused laser. Therefore we favor continuous streamlines throughout the entire channel expansion for in-line Raman measurements. In order to avoid the presence of circulating eddies, one can reduce the channel height h. Fig. 4 demonstrates both experimentally and numerically that a reduction of the channel height from h = 40 μm to h = 20 μm makes the circulating eddies disappear in case of an in-plane geometry with W/w = 5, L = w and w = 50 μm. The cell-free blood plasma domains at the reduced channel height in Fig. 4(a) have a large extent of about 50 μm by 50 μm enabling exclusive plasma domain optical sensing. On the other hand, the reduction of the channel height implies that the optical interaction length for Raman scattering is reduced. However, the channel height h = 20 μm is well suited for Raman experiments with a 60× air objective because the depth of the field is of the same order as the height of the microfluidic channel. The only drawback of too large plasma domains is the strongly reduced plasma velocity in the outer regions of the expansion compared to the center of the channel so that sample exchange here cannot be considered instantaneous.
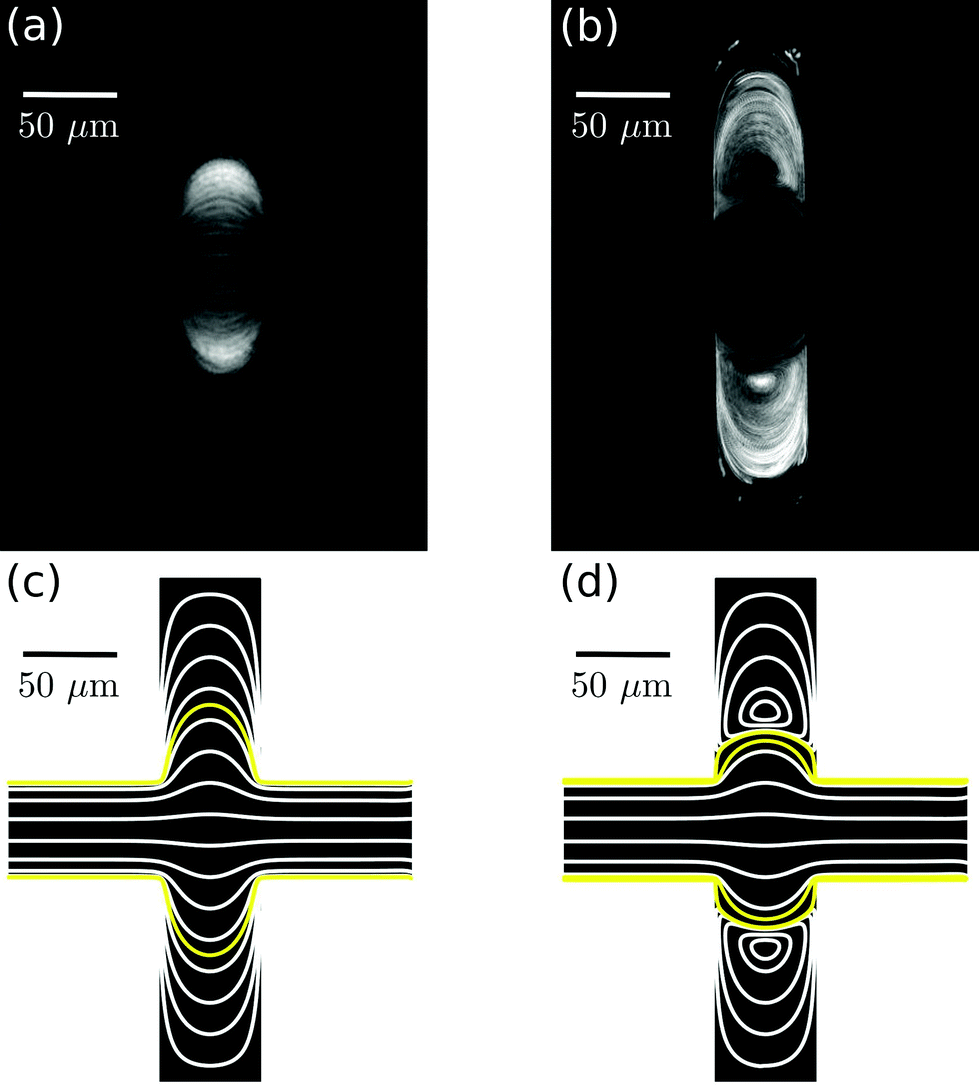 | ||
| Fig. 4 Different flow patterns for the same in-plane channel geometry, but different channel depths of h = 20 μm (a, c) and h = 40 μm (b, d). Panels (a–b) show the standard deviation of microfluidic experiments whereas panels (c–d) illustrate the streamlines of corresponding creeping flow simulations. | ||
The size of the cell-depleted domains is nearly constant at all underpressures we have applied in the range of 1–100 mbar. Only at low flow velocities at pressures below 10 mbar, we observe a tendency towards smaller-sized blood plasma domains. If no underpressure is applied to the outlet of the microchannel, blood stands still and the geometric channel expansion is entirely filled with red blood cells, including the corners. At non-zero flow velocities, cell-free domains appear which is due to the stable hydrodynamical effect. Apart from fluctuations at the boundary, the outline of cell-free domains is constant with respect to time. The averaged images of two seconds long time series in Fig. 3 demonstrate the temporal stability of flow patterns. In fact, it is possible to maintain the same flow pattern for several hours in our setup.
Raman hemoglobin evidence in bovine plasma
By choosing the Raman excitation wavelength of 408 nm, the incident light field is in resonance with part of a continuum of electronic transitions arising from the central and photoactive part of the hemoglobin molecule. The entire continuum is called the Soret band and becomes most apparent at about 420 nm in the hemoglobin absorption spectrum as shown for oxygenated hemoglobin in Fig. 5(a). The porphyrin structure of the heme group is highly Raman active, in particular, if both the resonant laser excitation and the Raman emission are located within its Soret band.24 We see strong Raman scattering by oxygenated hemoglobin, in particular at 1375 cm−1 which corresponds to an emission wavelength of about 432 nm considering 408 nm Raman excitation. The reference Raman spectrum of oxygenated hemoglobin is shown at the bottom of Fig. 5(b). In the following part, we focus on the Raman signature of oxygenated hemoglobin in liquid plasma solutions.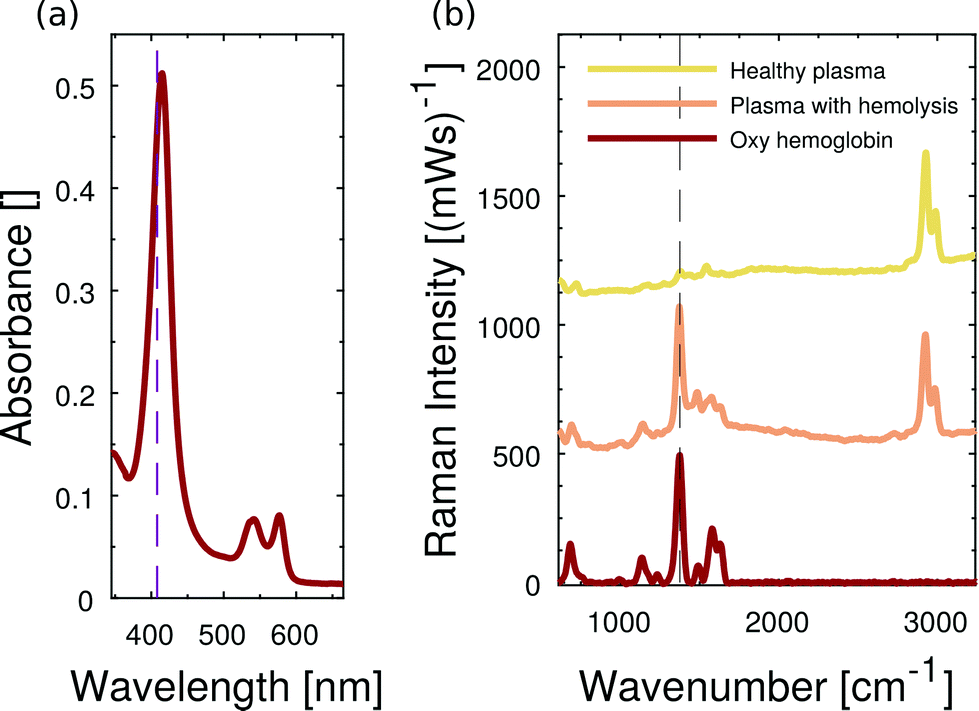 | ||
| Fig. 5 Resonance Raman spectroscopy of hemoglobin in liquids. (a) Absorption spectrum of oxyhemoglobin, the resonant laser excitation at 408 nm is spectrally located inside the Soret band. (b) Raman spectra of plasma without (yellow) and plasma with (orange) oxyhemoglobin in microfluidic channels. The most dominant Raman spectroscopic evidence of hemoglobin appears at 1375 cm−1. The distinctive PDMS signature is spectrally located at about 2900 cm−1. Spectra are vertically offset for clarity. | ||
The microfluidic channel is filled either with healthy bovine plasma or with bovine plasma that carries dissolved oxygenated hemoglobin. The acquired Raman spectra of both pure plasma and hemoglobin-enriched plasma are plotted in Fig. 5(b). Beside characteristic Raman scattering, we observe a continuous fluorescent background from bovine blood plasma proteins. In the case of added hemoglobin, however, the characteristic Raman signature of oxygenated hemoglobin appears and superimposes the fluorescent emission. The resonance Raman scattering from hemoglobin is so strong that it is not hidden in the fluorescent background. The double-shouldered Raman band at about 2900 cm−1 originates from Raman scattering by C–H bonds of the microfluidic chip material PDMS.
Quantitative Raman analysis of free hemoglobin in instantaneously created blood plasma
Firstly, we demonstrate that the microfluidic in-line plasma separation locally creates blood plasma as demanded. Here we prepare both a centrifuged blood plasma sample with 200 mg dl−1 dissolved free hemoglobin and a whole blood sample with 110 mg dl−1 dissolved free hemoglobin. Considering the hematocrit of that particular whole blood being 45%, the plasma phase of this second sample has an effective free hemoglobin concentration of 200 mg dl−1 as well. The Raman excitation is directed into the dynamically created cell-free plasma domain of the whole blood sample in the microfluidic flow cell, as illustrated in Fig. 2(a). The same optical alignment is used, when the conventionally centrifuged plasma sample flows through the microchannel. In Fig. 6(a) we compare the baseline corrected38 Raman spectra of these two samples and observe a perfect agreement. Furthermore, the equivalent concentration of free hemoglobin dissolved in water results in the same hemoglobin Raman peak intensity at 1375 cm−1. Minor differences between the latter and the plasma spectra – in particular in the spectral region at about 1530 cm−1 – are due to the presence of beta-carotene in blood plasma samples.14,29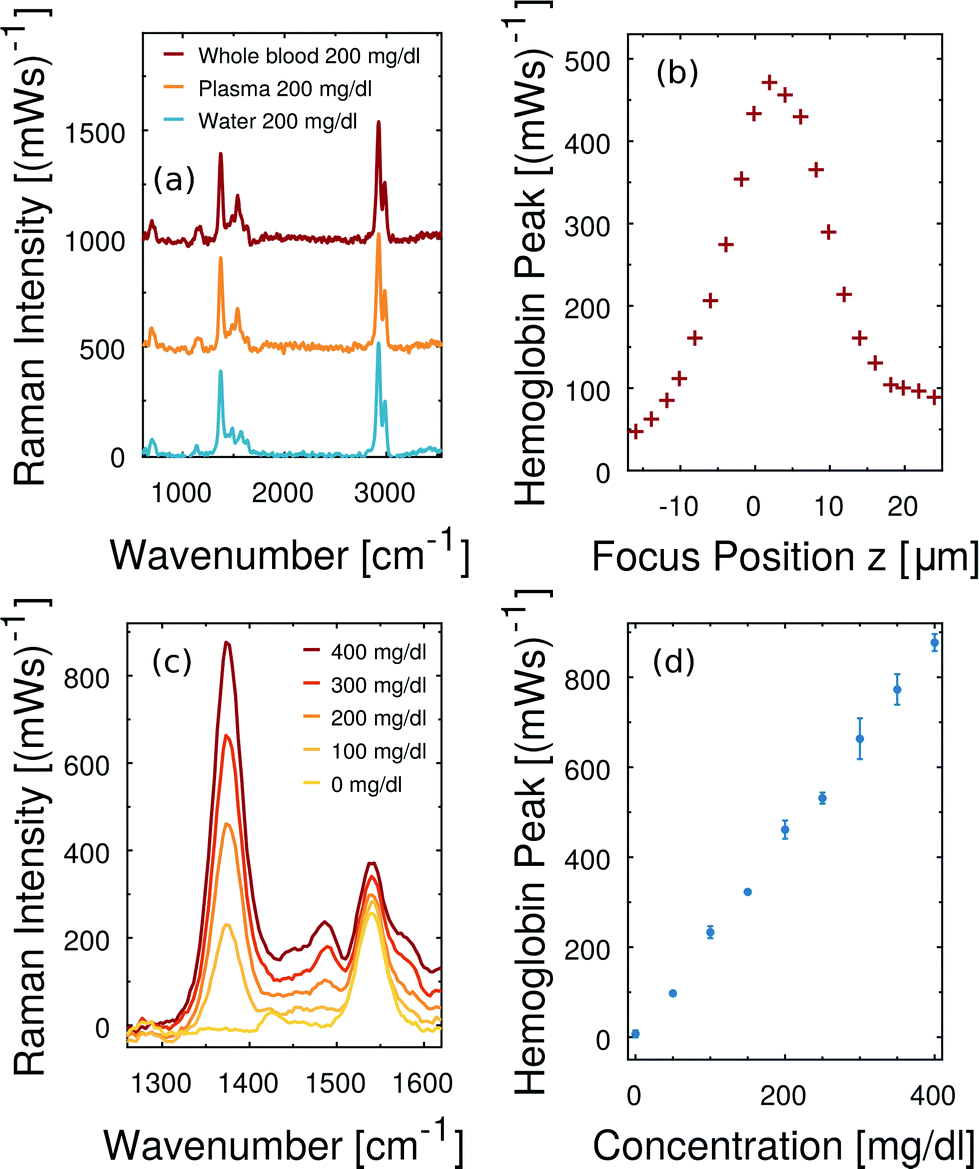 | ||
| Fig. 6 Free hemoglobin measurements in fractionated whole blood flow. (a) Baseline corrected Raman spectra of fractionated whole blood, blood plasma and water, all carrying dissolved free hemoglobin through the microchannel. The mentioned concentrations refer to free hemoglobin in the plasma/aqueous phase. (b) The hemoglobin Raman peak intensity is sensitive to the alignment of the optical system. (c) Averaged baseline-corrected Raman spectra at different hemoglobin concentrations. The characteristic hemoglobin peak increases monotonically with increasing free hemoglobin content. (d) Raman peak intensity scales linearly in the hemolysis range of clinical relevance. In all Raman measurements we apply 0.8 mW laser power at the sample and 3 seconds integration time. | ||
High consistency in hemoglobin Raman signal intensity enables quantitative free hemoglobin sensing. However, we note that reproducible scattering experiments are sensitive to the optical alignment. In particular, the small depth of field of our optical collection system and small channel depth imply that minor changes regarding the in-focus position z have a measurable effect on the detected Raman scattering. Fig. 6(b) illustrates the peak intensity of the smoothed hemoglobin signal at 1375 cm−1 as a function of the focus position z. The channel is 20 μm deep. The collected Raman intensity is not constant across this range demonstrating how important a stable position of the microfluidic channel with respect to the optical system is in order to quantitatively measure Raman scattering intensity in microfluidics.
In particular due to the careful sample exchange at the microfluidic inlet we can guarantee a stable optical alignment throughout a series of Raman measurements. We perform instantaneous blood plasma fractionation with the prepared whole blood samples of known free hemoglobin concentrations. Confocal Raman excitation is directed to the cell-free plasma domain in the microfluidic whole blood flow. As the free hemoglobin concentration of whole blood varies, the Raman intensity of the prominent hemoglobin peak at 1375 cm−1 in Fig. 6(c) changes. The smoothed Raman peak intensity after the baseline correction scales monotonically with the hemoglobin concentration. Fig. 6(d) shows a linear scaling and high reproducibility at all analyzed concentrations. Each error bar results from three independent Raman measurements on dynamically fractionated blood plasma at the same hemoglobin concentration. Accordingly, free hemoglobin concentrations of unknown whole blood samples can be quantified. Fig. 6(c and d) show that there is a negligible offset in terms of free hemoglobin which is natural in any whole blood sample. It means, furthermore, that no in vitro hemolysis occurs during our microfluidic blood experiments.
Fig. 7 illustrates the applied baseline correction method and determines the level of detection (LoD) for free hemoglobin in blood plasma by utilizing the derived concentration curve from Fig. 6(d). For baseline correction of raw spectra we employ asymmetrically reweighted penalized least squares smoothing,38 thereby achieving a symmetric noise distribution on both sides of the baseline. Throughout our study we apply the algorithm provided by Baek et al.38 with the parameter λ = 107. The baseline corrected spectrum is shown in Fig. 7(b) and its corresponding histogram is shown in Fig. 7(c). The LoD of the hemoglobin peak at 1375 cm−1 is defined as three times as high as the standard deviation σ of the noise distribution around zero. This domination compared to the noise ensures that the hemoglobin peak is measurable. From Fig. 7(b) we extract 3σ = 52 (mW)−1 which means a LoD of 22 mg dl−1.
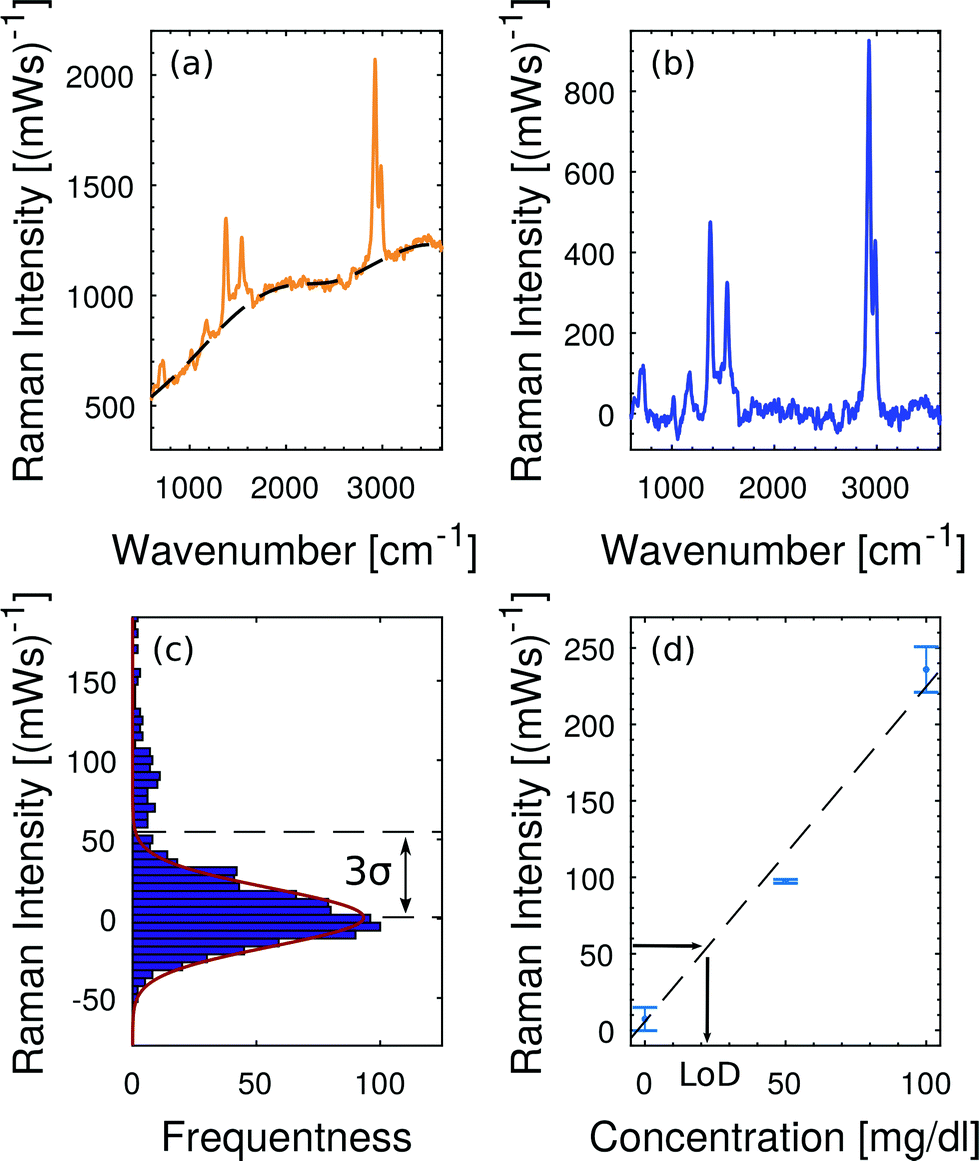 | ||
| Fig. 7 Baseline fitting and level of detection (LoD). (a) Raw spectrum at 200 mg dl−1 hemoglobin in plasma and the corresponding baseline fit38 which accounts for the fluorescent background. (b) Baseline-corrected Raman spectrum. (c) Noise distribution close to the baseline fit. The LoD is defined as 3σ of this noise distribution. (d) The concentration calibration from Fig. 6(d) translates 3σ = 52 (mW)−1 into a LoD of 22 mg dl−1. | ||
Near-infrared Raman spectroscopy of fractionated plasma in the microfluidic whole blood flow
Raman scattering from other plasma molecules than resonantly excited hemoglobin and beta-carotene is hidden in the fluorescent background while using the 408 nm laser. In order to detect weaker Raman contributions we aim to avoid fluorescence and apply near-infrared excitation at 785 nm with 70 mW of laser power at the sample. The Raman spectra of blood plasma at near-infrared excitation were starting points in several PLS regression studies15,16,39 which calibrate multi-component models to quantify a set of blood parameters like glucose, cholesterol, triglyceride, urea, total protein and albumin simultaneously from a single Raman spectrum.In addition to the general problems of fluorescence and inherent shot noise in liquid plasma samples, we face the problem of strong Raman background from transparent PDMS, our choice of microfluidic host material. The amplitude of this background in microfluidic plasma measurements becomes apparent in Fig. 8(a). In contrast, the intensity of Raman and fluorescent scattering from blood plasma in the channel is weaker. But the Raman spectrum shows small characteristic Raman features of blood plasma. In order to restore the pure blood plasma Raman spectrum, we apply a scaled subtraction of the spectrum by the PDMS background spectrum.
 | ||
| Fig. 8 NIR Raman spectroscopy of blood plasma in a microfluidic channel with 3 minutes integration time. (a) Fluorescent and Raman scattering from blood plasma are superimposed by the out-off focus Raman scattering by PDMS. (b) A scaled subtraction of the PDMS Raman signature from the spectrum corrects for the PDMS background and restores the pure spectrum of blood plasma. | ||
The result of this scaled subtraction is plotted in Fig. 8(b), next to the pure plasma spectrum that was recorded from volume without the PDMS channel. The spectra match qualitatively in terms of fluorescent background and characteristic Raman lines. Minor differences at prominent PDMS wavenumbers are artifacts of the scaled subtraction routine. Nevertheless, the spectrum is very similar to those reported in previous studies.15,17,39 In particular, the strong peak of phenylalanine near 1000 cm−1 as well as the peaks at 1140 cm−1 (glucose) and 1450 cm−1 (arrows in Fig. 8(c)) are in good agreement.
The intensity of our regular blood plasma Raman spectrum at near-infrared excitation could be significantly enhanced by the addition of metallic nanoparticles utilizing the SERS effect. Feng et al.17,19 and Lin et al.20 compared the regular plasma spectrum to the one where plasma is mixed with silver/gold colloids. They demonstrated a multiple increase of Raman peak intensity, whereas background fluorescent decreased. These SER spectra serve then as a basis for cancer diagnostics. As our regular plasma spectrum is of comparable quality, we assume that the addition of metallic colloids would also enhance Raman scattering from plasma in our microfluidic channel enabling SERS classification studies – such as cancer diagnostics.
4 Conclusions
In this proof of principle study, we present a technology that enables microfluidic blood plasma monitoring for Raman accessible parameters at low blood consumption. We combine the instantaneous blood plasma domain creation in the microfluidic whole blood flow with Raman microscopy and demonstrate hemolysis detection at relevant concentrations as well as near-infrared Raman spectroscopy of plasma in microchannels.Much attention has been paid to liquid blood and plasma samples in large volumes like quartz cuvettes15,16,39 or Raman imaging at the cellular level.14,40,41 This work fits the intermediate interval of sizes in the several micron range where dynamical effects in blood flow occur. In our experiments the expansion of the cell-free plasma layer from about 1 μm to 10 μm or more is necessary in order to keep the cells unaffected by laser light and to match the capabilities of the micro-Raman system. The shape of the fractionated plasma domains can be tailored by modifying the geometry of the expansion. However, there is a tradeoff regarding the channel height h. In shallow channels circulating eddies are avoided. But at the same time, the optical interaction length for Raman scattering by sample volume inside the channel is reduced. Furthermore the detected Raman background intensity from the microfluidic chip material PDMS is less suppressed.
In our first application example we demonstrate high sensitivity and appreciable linearity in free hemoglobin sensing by means of resonance Raman spectroscopy. The excitation wavelength has been chosen in order to specifically probe for hemoglobin. Fortunately, the Raman intensity from hemoglobin is strong enough to overcome the fluorescent background. This method enables instantaneous hemolysis detection on whole blood.
In our second application example we focus on the much weaker Raman scattering from microfluidic blood plasma domains at near-infrared excitation. The background Raman intensity from PDMS dominates over the plasma signal. However, the scaled subtraction of the background spectrum allows the restoration of the pure plasma signature. Mixtures of blood plasma with metallic colloids show enhanced Raman scattering compared to regular Raman spectra which we reproduce in the microfluidic setting. Such mixtures have potential for example in cancer diagnostics19,20 and can be applied in microfluidics in order to reduce both colloid and blood consumption.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Innovation Fund Denmark with the project no. 5106-00015B, HemoPoC. We thank the team at Radiometer for regular bovine blood delivery and fruitful discussions. We thank Chen Zhou and Uriel Levy for fruitful discussions.References
- T. Secomb, Symp. Soc. Exp. Biol., 1995, 49, 305–321 CAS.
- A. R. Pries, T. Secomb and P. Gaehtgens, Cardiovasc. Res., 1996, 32, 654–667 CrossRef CAS PubMed.
- D. A. Fedosov, B. Caswell, A. S. Popel and G. E. M. Karniadakis, Microcirculation, 2010, 17, 615–628 CrossRef PubMed.
- T. G. Spiro and T. C. Streakas, Biochim. Biophys. Acta, 1972, 263, 830–833 Search PubMed.
- M. Faivre, M. Abkarian, K. Bickraj and H. A. Stone, Biorheology, 2006, 43, 147–159 Search PubMed.
- E. Sollier, M. Cubizolles, Y. Fouillet and J. L. Achard, Biomed. Microdevices, 2010, 12, 485–497 CrossRef PubMed.
- S. Tripathi, Y. V. B. Varun Kumar, A. Prabhakar, S. S. Joshi and A. Agrawal, J. Micromech. Microeng., 2015, 25, 083001 CrossRef.
- S. Tripathi, Y. V. B. Kumar, A. Agrawal, A. Prabhakar and S. S. Joshi, Sci. Rep., 2016, 6, 26749 CrossRef CAS PubMed.
- A. F. Chrimes, K. Khoshmanesh, P. R. Stoddart, A. Mitchell and K. Kalantar-Zadeh, Chem. Soc. Rev., 2013, 42, 5880–5906 RSC.
- S.-A. Leung, R. F. Winkle, R. C. R. Wootton and A. J. deMello, Analyst, 2005, 130, 46–51 RSC.
- P. C. Ashok, G. P. Singh, H. a. Rendall, T. F. Krauss and K. Dholakia, Lab Chip, 2011, 11, 1262–1270 RSC.
- S. H. Yazdi and I. M. White, Biomicrofluidics, 2012, 6, 1–9 Search PubMed.
- I. J. Jahn, O. Žukovskaja, X.-S. Zheng, K. Weber, T. W. Bocklitz, D. Cialla-May and J. Popp, Analyst, 2017, 142, 1022–1047 RSC.
- C. G. Atkins, K. Buckley, M. W. Blades and R. F. Turner, Appl. Spectrosc., 2017, 71, 767–793 CrossRef CAS PubMed.
- A. J. Berger, T.-W. Koo, I. Itzkan, G. Horowitz and M. S. Feld, Appl. Opt., 1999, 38, 2916–2926 CrossRef CAS PubMed.
- A. M. K. Enejder, T.-W. Koo, J. Oh, M. Hunter, S. Sasic and M. S. Feld, Opt. Lett., 2002, 27, 2004–2006 CrossRef CAS PubMed.
- S. Feng, R. Chen, J. Lin, J. Pan, G. Chen, Y. Li, M. Cheng, Z. Huang, J. Chen and H. Zeng Haishan, Biosens. Bioelectron., 2010, 25, 2414–2419 CrossRef CAS PubMed.
- S. Feng, R. Chen, J. Lin, J. Pan, Y. Wu, Y. Li, J. Chen and H. Zeng, Biosens. Bioelectron., 2011, 26, 3167–3174 CrossRef CAS PubMed.
- S. Feng, D. Lin, J. Lin, B. Li, Z. Huang, G. Chen, W. Zhang, L. Wang, J. Pan, R. Chen and H. Zeng, Analyst, 2013, 138, 3967–3974 RSC.
- D. Lin, J. Pan, H. Huang, G. Chen, S. Qiu, H. Shi, W. Chen, Y. Yu, S. Feng and R. Chen, Sci. Rep., 2014, 4, 1–8 Search PubMed.
- A. Bonifacio, S. Cervo and V. Sergo, Anal. Bioanal. Chem., 2015, 407, 8265–8277 CrossRef CAS PubMed.
- A. Bonifacio, S. Dalla Marta, R. Spizzo, S. Cervo, A. Steffan, A. Colombatti and V. Sergo, Anal. Bioanal. Chem., 2014, 406, 2355–2365 CrossRef CAS PubMed.
- EDQM, Guide to the preparation, use and quality assurance of blood components, Recommendation No. R (95) 15, 2015 Search PubMed.
- B. B. Johnson and W. L. Peticolas, Annu. Rev. Phys. Chem., 1976, 27, 465–491 CrossRef CAS.
- D. L. Rousseau, J. M. Friedman and P. F. Williams, in The Resonance Raman Effect, ed. A. Weber, Springer Berlin Heidelberg, Berlin, Heidelberg, 1979, pp. 203–252 Search PubMed.
- H. Xu, E. J. Bjerneld, M. Käll and M. Börjesson, Phys. Rev. Lett., 1999, 83, 4357–4360 CrossRef CAS.
- I. P. T. Filho, J. Terner, R. N. Pittman and K. R. Ward, Am. J. Physiol.: Heart Circ. Physiol., 2005, 289, 488–495 CrossRef PubMed.
- V. Sikirzhytski, A. Sikirzhytskaya and I. K. Lednev, Appl. Spectrosc., 2011, 65, 1223–1232 CrossRef CAS PubMed.
- M. Casella, A. Lucotti, M. Tommasini, M. Bedoni, E. Forvi, F. Gramatica and G. Zerbi, Spectrochim. Acta, Part A, 2011, 79, 915–919 CrossRef CAS PubMed.
- L. Rimai, I. Salmeen and D. H. Petering, Biochemistry, 1975, 14, 378–382 CrossRef CAS PubMed.
- M. Lu, L. Zhao, Y. Wang, G. You, X. Kan, Y. Zhang, N. Zhang, B. Wang, Y.-J. Guo and H. Zhou, Artif. Cells, Nanomed., Biotechnol., 2014, 42, 63–69 CrossRef CAS PubMed.
- L. T. Kerr, H. J. Byrne and B. M. Hennelly, Anal. Methods, 2015, 7, 5041–5052 RSC.
- K. Chau, B. Millare, A. Lin, S. Upadhyayula, V. Nunez, H. Xu and V. I. Vullev, Microfluid. Nanofluid., 2011, 10, 907–917 CrossRef CAS.
- C. Conti, M. Realini, C. Colombo, K. Sowoidnich, N. K. Afseth, M. Bertasa, A. Botteon and P. Matousek, Anal. Chem., 2015, 87, 5810–5815 CrossRef CAS PubMed.
- Y. Cho, S. W. Song, J. Sung, Y.-S. Jeong, C. R. Park and H. M. Kim, Analyst, 2017, 142, 3613–3619 RSC.
- COMSOL Inc., COMSOL Multiphysics Reference Manual, version 5.3a, 2018 Search PubMed.
- H. Moffat, J. Fluid Mech., 1964, 18, 1–18 CrossRef.
- S.-J. Baek, A. Park, Y.-J. Ahn and J. Choo, Analyst, 2015, 140, 250–257 RSC.
- D. Rohleder, W. Kiefer and W. Petrich, Analyst, 2004, 129, 906 RSC.
- A. Ramoji, U. Neugebauer, T. Bocklitz, M. Foerster, M. Kiehntopf, M. Bauer and J. Popp, Anal. Chem., 2012, 84, 5335–5342 CrossRef CAS PubMed.
- J. Dybas, M. Grosicki, M. Baranska and K. M. Marzec, Analyst, 2018, 143, 3489–3498 RSC.
| This journal is © The Royal Society of Chemistry 2019 |