
Pushing the limits of detection for proteins secreted from single cells using quantum dots†
 abc
and
Jered B.
Haun
*abde
abc
and
Jered B.
Haun
*abde
Abstract
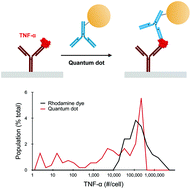
Single cell analysis methods are increasingly being utilized to investigate how individual cells process information and respond to diverse stimuli. Soluble proteins play a critical role in controlling cell populations and tissues, but directly monitoring secretion is technically challenging. Microfabricated well arrays have been developed to assess secretion at the single cell level, but these systems are limited by low detection sensitivity. Semiconductor quantum dots (QD) exhibit remarkably bright and photostable luminescence signal, but to date they have not been evaluated in single cell secretion studies using microfabricated well arrays. Here, we used QDs in a sandwich immunoassay to detect secretion of the soluble cytokine tumor necrosis factor-α (TNF-α) from single cells. To enhance detection sensitivity, we employed two different strategies. First, we used a unique single QD imaging approach, which provided a detection threshold (180 attomolar) that was >100-fold lower than previously reported results using QDs. We also amplified QD binding to each captured TNF-α molecule using the bioorthogonal cycloaddition reaction between trans-cyclooctene and tetrazine, which further lowered detection threshold to 60 attomolar. This is 6 orders of magnitude more sensitive than organic fluorophores that have been used for single cell secretion studies, and far surpasses single molecule resolution within sub-picoliter microwells that are used to assess single cell secretion. Finally, single cell secretion studies were performed using phorbol 12-myristate 13-acetate (PMA) differentiated and lipopolysaccharide (LPS) activated U-937 cells. TNF-α secretion was detected from 3-fold more single cells using the QD-based method in comparison to rhodamine, which was accomplished by extending sensitivity into the range of ∼2 to 10 000 molecules captured per microwell. In future work, we will apply this technique to assess immune cell secretion dynamics under diverse stimuli and disease settings. We will also incorporate multiplexing capabilities to evaluate the secretome at the resolution of single molecules.
- This article is part of the themed collection: Next wave advances in single cell analyses
 Please wait while we load your content...
Please wait while we load your content...

